Institute of Bioengineering, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Swiss Institute of Bioinformatics (SIB), Lausanne, Switzerland.
PLoS Comput Biol. 2018 Nov 19;14(11):e1006623. doi: 10.1371/journal.pcbi.1006623. eCollection 2018 Nov.
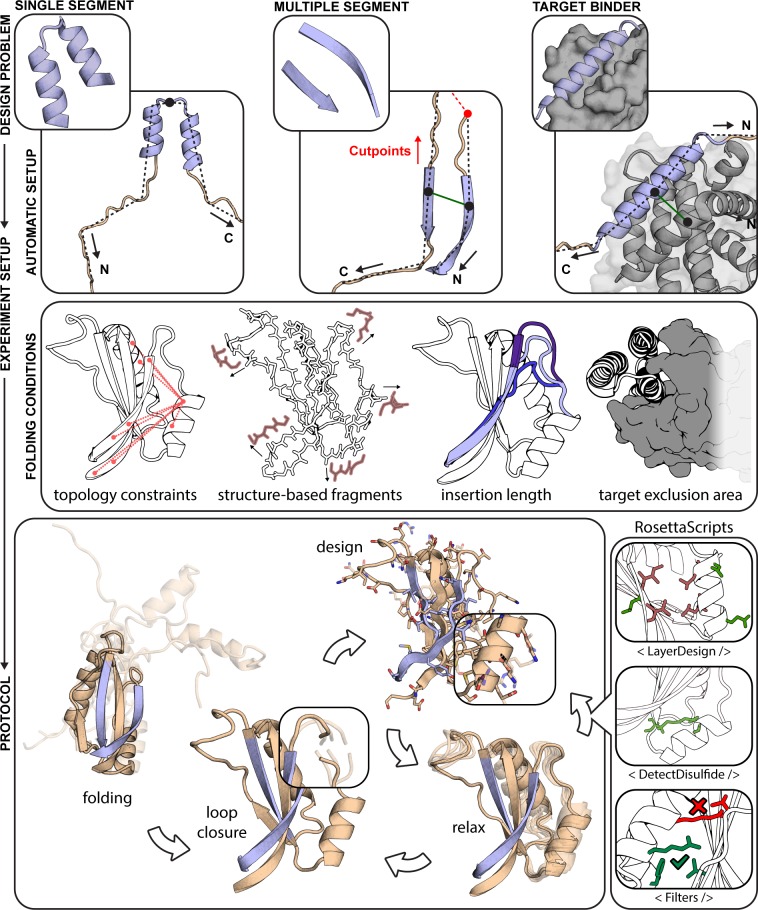
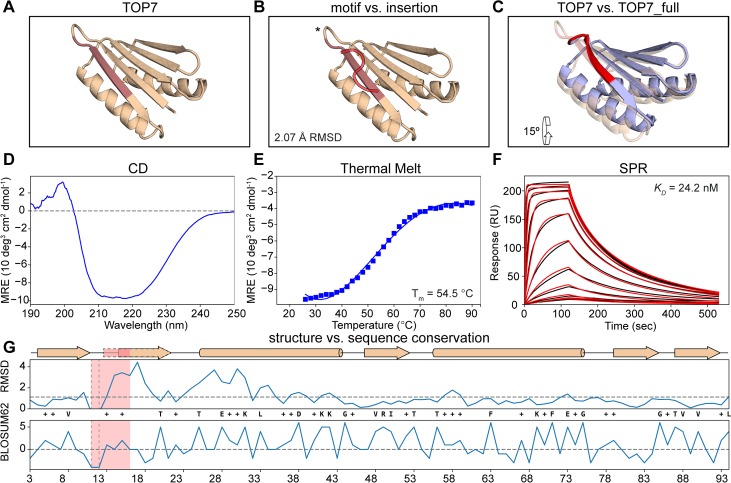
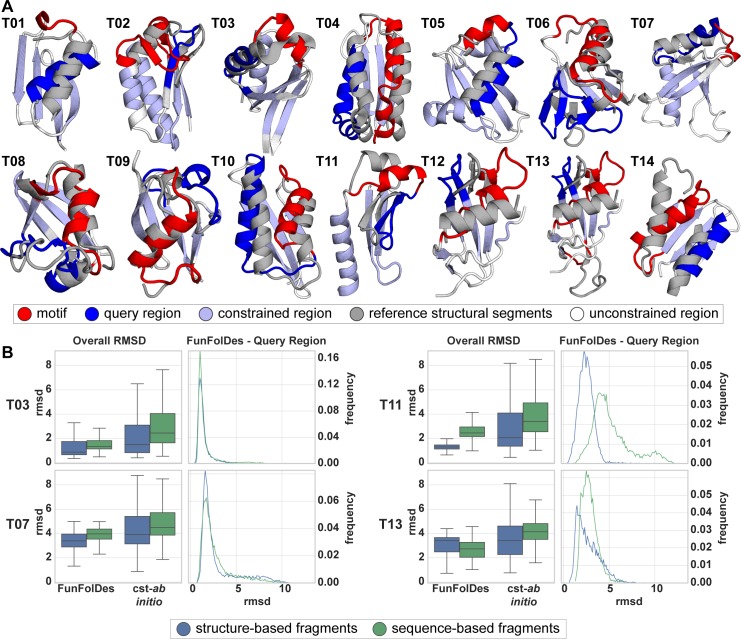
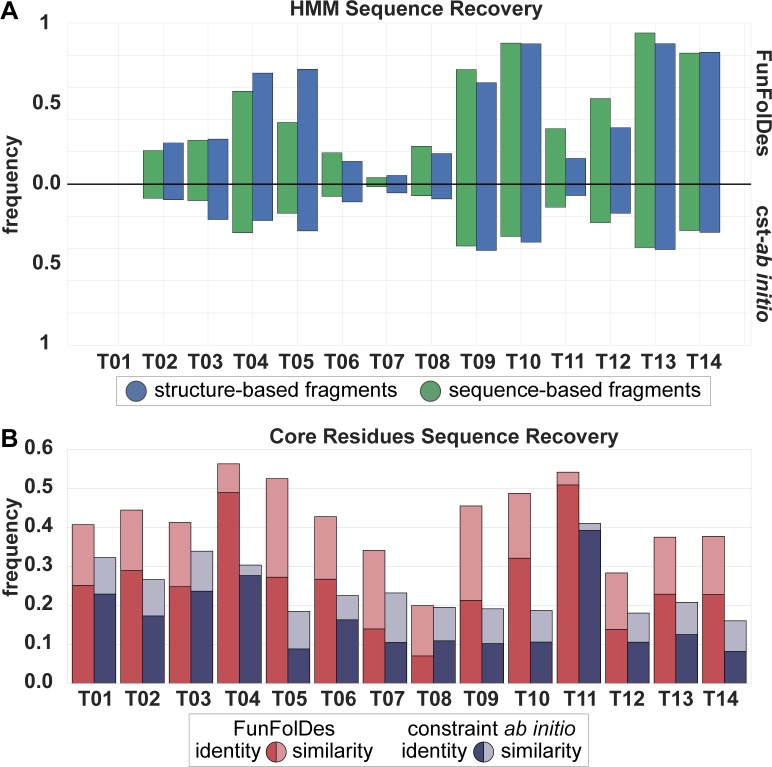
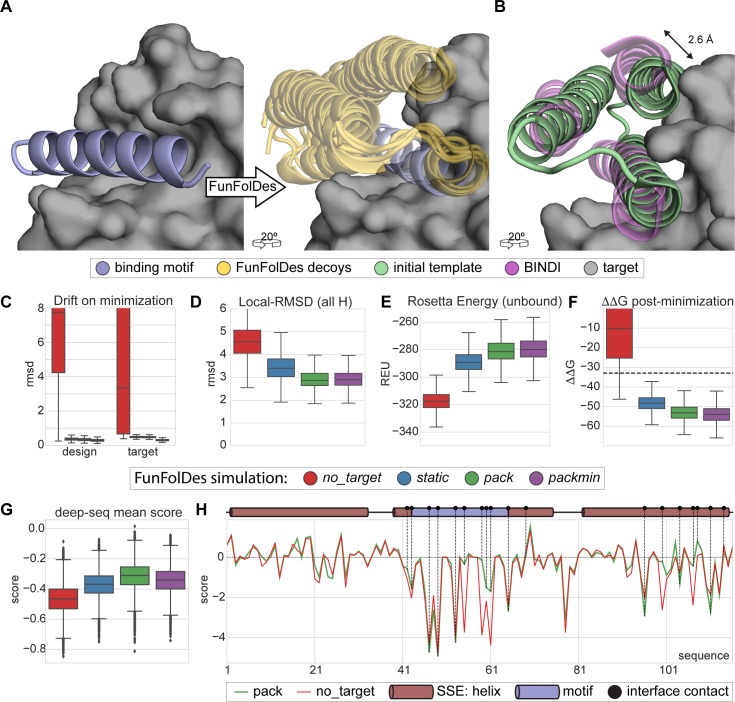
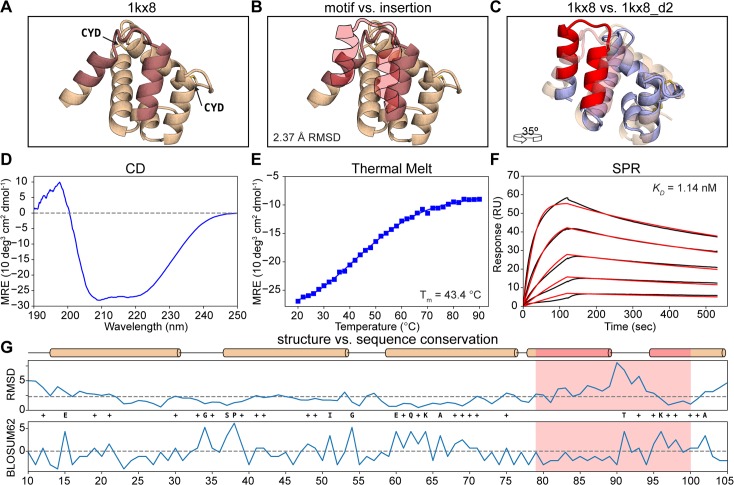
The robust computational design of functional proteins has the potential to deeply impact translational research and broaden our understanding of the determinants of protein function and stability. The low success rates of computational design protocols and the extensive in vitro optimization often required, highlight the challenge of designing proteins that perform essential biochemical functions, such as binding or catalysis. One of the most simplistic approaches for the design of function is to adopt functional motifs in naturally occurring proteins and transplant them to computationally designed proteins. The structural complexity of the functional motif largely determines how readily one can find host protein structures that are "designable", meaning that are likely to present the functional motif in the desired conformation. One promising route to enhance the "designability" of protein structures is to allow backbone flexibility. Here, we present a computational approach that couples conformational folding with sequence design to embed functional motifs into heterologous proteins-Rosetta Functional Folding and Design (FunFolDes). We performed extensive computational benchmarks, where we observed that the enforcement of functional requirements resulted in designs distant from the global energetic minimum of the protein. An observation consistent with several experimental studies that have revealed function-stability tradeoffs. To test the design capabilities of FunFolDes we transplanted two viral epitopes into distant structural templates including one de novo "functionless" fold, which represent two typical challenges where the designability problem arises. The designed proteins were experimentally characterized showing high binding affinities to monoclonal antibodies, making them valuable candidates for vaccine design endeavors. Overall, we present an accessible strategy to repurpose old protein folds for new functions. This may lead to important improvements on the computational design of proteins, with structurally complex functional sites, that can perform elaborate biochemical functions related to binding and catalysis.
功能蛋白的稳健计算设计有可能对转化研究产生深远影响,并拓宽我们对蛋白质功能和稳定性决定因素的理解。计算设计方案的成功率低,而且通常需要广泛的体外优化,这突出了设计能够执行重要生化功能(如结合或催化)的蛋白质的挑战性。设计功能的最简单方法之一是采用天然存在的蛋白质中的功能基序,并将其移植到计算设计的蛋白质中。功能基序的结构复杂性在很大程度上决定了人们是否能够轻易找到“可设计”的宿主蛋白结构,也就是说,这些结构可能以所需的构象呈现功能基序。增强蛋白质结构“可设计性”的一种有前途的途径是允许骨架灵活性。在这里,我们提出了一种计算方法,该方法将构象折叠与序列设计相结合,将功能基序嵌入异源蛋白质中 - Rosetta 功能折叠和设计(FunFolDes)。我们进行了广泛的计算基准测试,观察到执行功能要求会导致设计远离蛋白质的全局能量最小值。这一观察结果与几项实验研究一致,这些研究揭示了功能-稳定性权衡。为了测试 FunFolDes 的设计能力,我们将两个病毒表位移植到包括从头开始的“无功能”折叠在内的远距离结构模板中,这代表了出现可设计性问题的两个典型挑战。实验表征设计的蛋白质显示出与单克隆抗体的高结合亲和力,使它们成为疫苗设计工作的有价值候选物。总的来说,我们提出了一种可访问的策略,可以为新功能重新利用旧的蛋白质折叠。这可能会导致与结合和催化等复杂生化功能相关的具有复杂结构功能位点的蛋白质的计算设计得到重要改进。