Department of Neurology, Division of Neurobiology, Medical University of Innsbruck, Innsbruck, Austria.
MODAG GmbH, Wendelsheim, Germany.
Mov Disord. 2019 Feb;34(2):255-263. doi: 10.1002/mds.27562. Epub 2018 Nov 19.
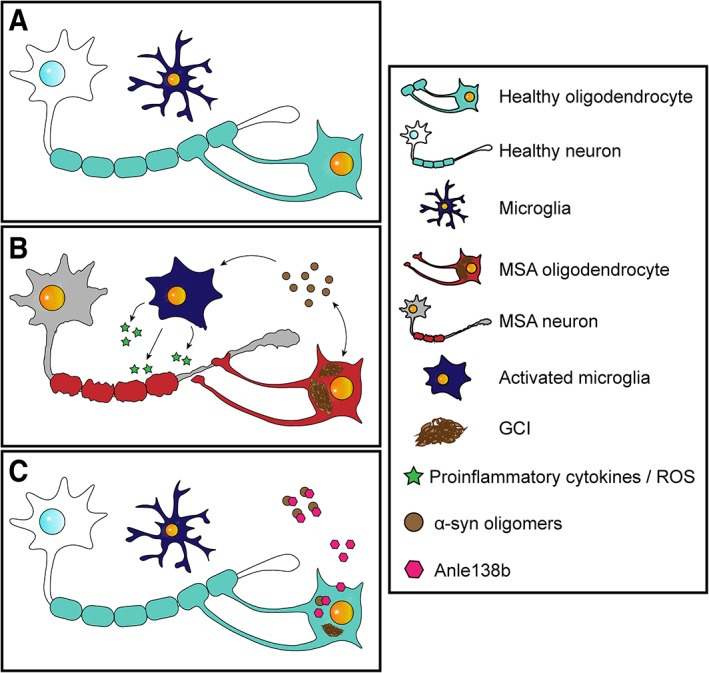
MSA is a fatal neurodegenerative disease characterized by autonomic failure and severe motor impairment. Its main pathological hallmark is the accumulation of α-synuclein in oligodendrocytes, leading to glial and neuronal dysfunction and neurodegeneration. These features are recapitulated in the PLP-hαSyn mouse model expressing human α-synuclein in oligodendrocytes. At present, there is no effective disease-modifying therapy. Previous experiments have shown that the aggregation inhibitor, anle138b, reduces neurodegeneration and behavioral deficits in mouse models of other proteinopathies.
To test the therapeutic potential of anle138b in a mouse model of MSA.
Two-month-old PLP-hαSyn mice were fed over a period of 4 months with pellets containing anle138b at two different doses (0.6 and 2 g/kg) and compared to healthy controls and PLP-hαSyn mice fed with placebo pellets. At the end of the treatment, behavioral and histological analyses were performed.
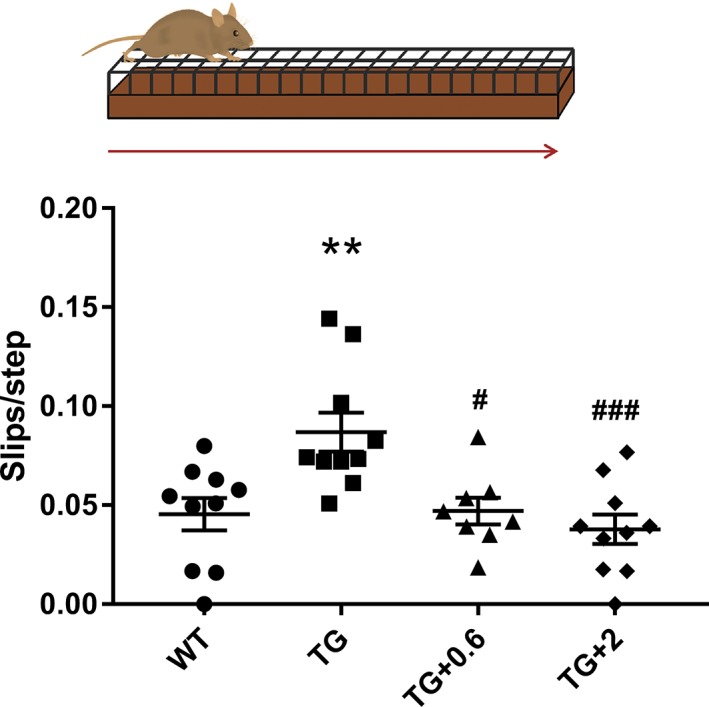
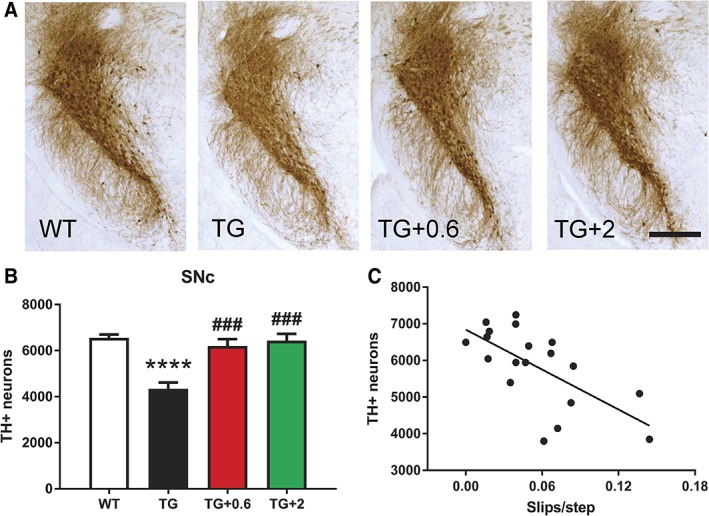
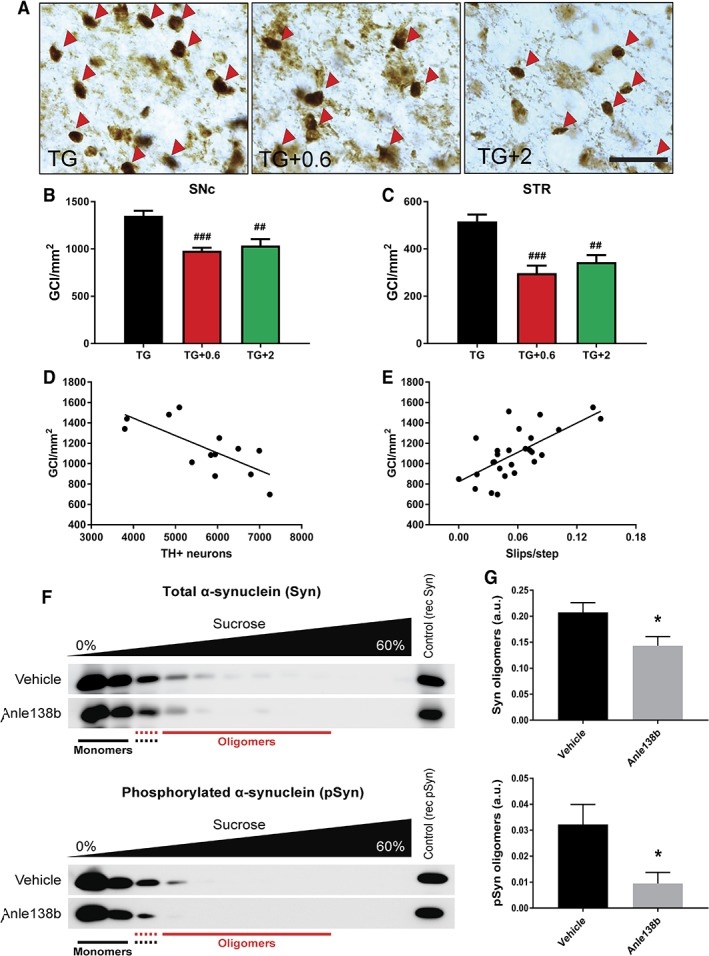
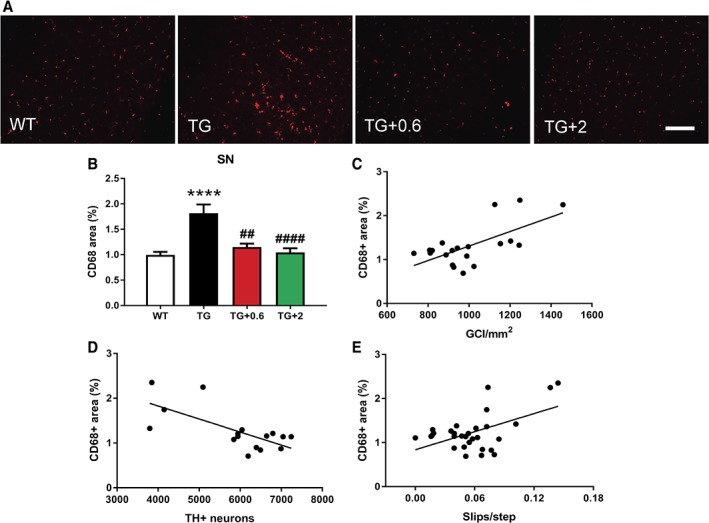
We observed a reversal of motor function to healthy control levels when PLP-hαSyn mice were treated with both doses of anle138b. Histological and molecular analyses showed a significant reduction in α-synuclein oligomers and glial cytoplasmic inclusions in animals fed with anle138b compared to nontreated mice. These animals also present preservation of dopaminergic neurons and reduction in microglial activation in SN correlating with the α-synuclein reduction observed.
Anle138b reduces α-synuclein accumulation in PLP-hαSyn mice, leading to neuroprotection, reduction of microglial activation, and preservation of motor function supporting the use of anle138b in a future clinical trial for MSA. © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
MSA 是一种致命的神经退行性疾病,其特征是自主功能衰竭和严重的运动障碍。其主要病理标志是α-突触核蛋白在少突胶质细胞中的积累,导致神经胶质和神经元功能障碍和神经退行性变。这些特征在表达人α-突触核蛋白的 PLP-hαSyn 小鼠模型中得到了重现。目前,尚无有效的疾病修饰疗法。先前的实验表明,聚集抑制剂 anle138b 可减少其他蛋白病变模型小鼠的神经退行性变和行为缺陷。
测试 anle138b 在 MSA 小鼠模型中的治疗潜力。
用含有 anle138b 的两种不同剂量(0.6 和 2 g/kg)的小丸喂养 2 个月大的 PLP-hαSyn 小鼠,持续 4 个月,并与健康对照和用安慰剂小丸喂养的 PLP-hαSyn 小鼠进行比较。治疗结束时进行行为和组织学分析。
当用两种剂量的 anle138b 治疗 PLP-hαSyn 小鼠时,我们观察到其运动功能恢复到健康对照水平。与未治疗的小鼠相比,用 anle138b 喂养的动物的α-突触核蛋白寡聚物和神经胶质细胞质内含物明显减少。这些动物还表现出多巴胺能神经元的保存和 SN 中小胶质细胞激活的减少,与观察到的α-突触核蛋白减少相关。
Anle138b 减少了 PLP-hαSyn 小鼠中α-突触核蛋白的积累,导致神经保护、小胶质细胞激活减少和运动功能的保留,支持在未来的 MSA 临床试验中使用 anle138b。