School of Biotechnology, Amrita Vishwa Vidyapeetham, Kollam, Kerala, India.
Department of Mathematics and Statistics, Villanova University, Villanova, PA, USA.
BMC Genomics. 2018 Nov 21;19(1):835. doi: 10.1186/s12864-018-5231-7.
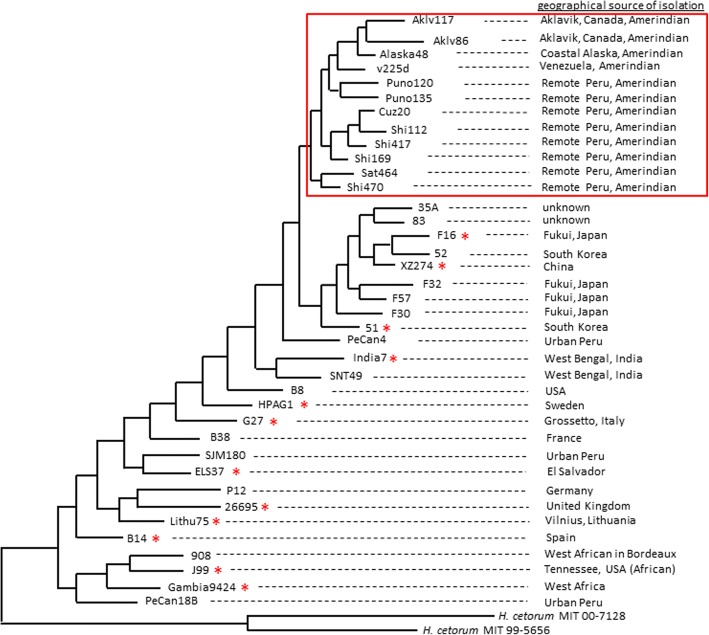
Helicobacter pylori is a human stomach pathogen, naturally-competent for DNA uptake, and prone to homologous recombination. Extensive homoplasy (i.e., phylogenetically-unlinked identical variations) observed in H. pylori genes is considered a hallmark of such recombination. However, H. pylori also exhibits a high mutation rate. The relative adaptive role of homologous recombination and mutation in species diversity is a highly-debated issue in biology. Recombination results in homoplasy. While convergent mutation can also account for homoplasy, its contribution is thought to be minor. We demonstrate here that, contrary to dogma, convergent mutation is a key contributor to Helicobacter pylori homoplasy, potentially driven by adaptive evolution of proteins.
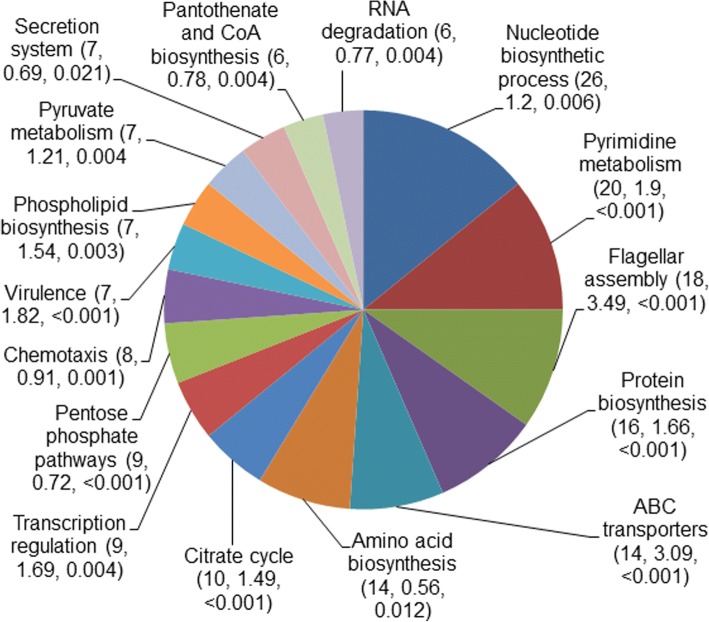
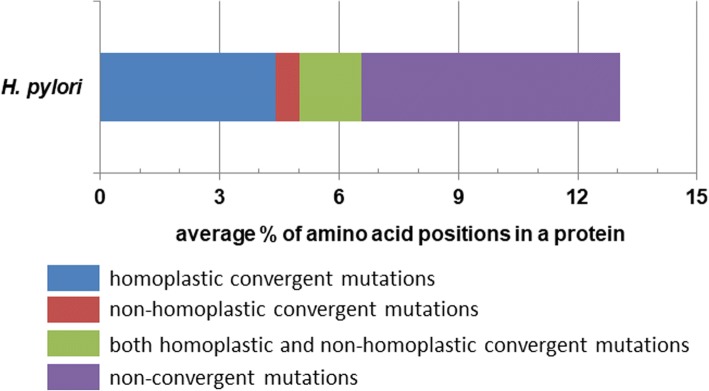
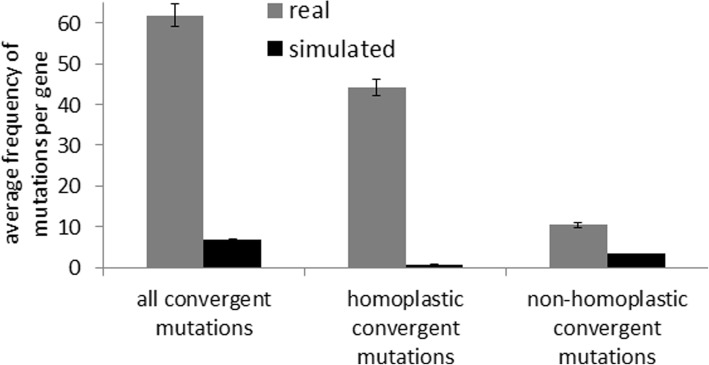
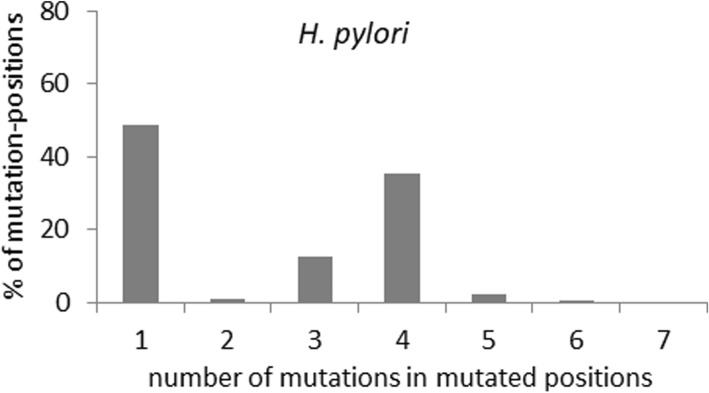
Our present genome-wide analysis shows that homoplastic nonsynonymous (amino acid replacement) changes are not typically accompanied by homoplastic synonymous (silent) variations. Moreover, the majority of the codon positions with homoplastic nonsynonymous changes also contain different (i.e. non-homoplastic) nonsynonymous changes arising from mutation only. This indicates that, to a considerable extent, nonsynonymous homoplasy is due to convergent mutations. High mutation rate or limited availability of evolvable sites cannot explain this excessive convergence, as suggested by our simulation studies. Rather, the genes with convergent mutations are overrepresented in distinct functional categories, suggesting possible selective responses to conditions such as distinct micro-niches in single hosts, and to differences in host genotype, physiology, habitat and diet.
We propose that mutational convergence is a key player in H. pylori's adaptation and extraordinary persistence in human hosts. High frequency of mutational convergence could be due to saturation of evolvable sites capable of responding to selection pressures, while the number of mutable residues is far from saturation. We anticipate a similar scenario of mutational vs. recombinational genome dynamics or plasticity for other naturally competent microbes where strong positive selection could favor frequent convergent mutations in adaptive protein evolution.
幽门螺杆菌是一种人类胃部病原体,能够自然地摄取 DNA,并且易于发生同源重组。在幽门螺杆菌基因中观察到广泛的同型(即系统发育上无关联的相同变异)被认为是这种重组的标志。然而,幽门螺杆菌也表现出高突变率。同源重组和突变在物种多样性中的相对适应性作用是生物学中一个备受争议的问题。重组导致同型,而趋同突变也可以解释同型,但认为其贡献较小。我们在这里证明,与传统观点相反,趋同突变是幽门螺杆菌同型的一个关键贡献因素,可能是由蛋白质的适应性进化驱动的。
我们目前的全基因组分析表明,同型非同义(氨基酸替换)变化通常不伴有同型同义(沉默)变化。此外,大多数具有同型非同义变化的密码子位置也包含仅由突变引起的不同(即非同型)非同义变化。这表明,在相当大的程度上,非同义同型是由于趋同突变所致。高突变率或可进化位点的有限可用性不能解释这种过度趋同,正如我们的模拟研究所表明的那样。相反,具有趋同突变的基因在不同的功能类别中过度表达,这表明可能存在对宿主内不同微生境等条件的选择性反应,以及宿主基因型、生理、栖息地和饮食的差异。
我们提出,趋同突变是幽门螺杆菌适应和在人类宿主中非凡持久性的关键因素。趋同突变的高频可能是由于能够响应选择压力的可进化位点饱和,而可突变残基的数量远未饱和。我们预计在其他自然有能力的微生物中也会出现类似的突变与重组基因组动态或可塑性的情况,其中强烈的正选择可能有利于适应性蛋白进化中的频繁趋同突变。