Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC-JA), Facultad de Ciencias Experimentales (Área de Genética), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
Genes (Basel). 2021 Mar 6;12(3):377. doi: 10.3390/genes12030377.
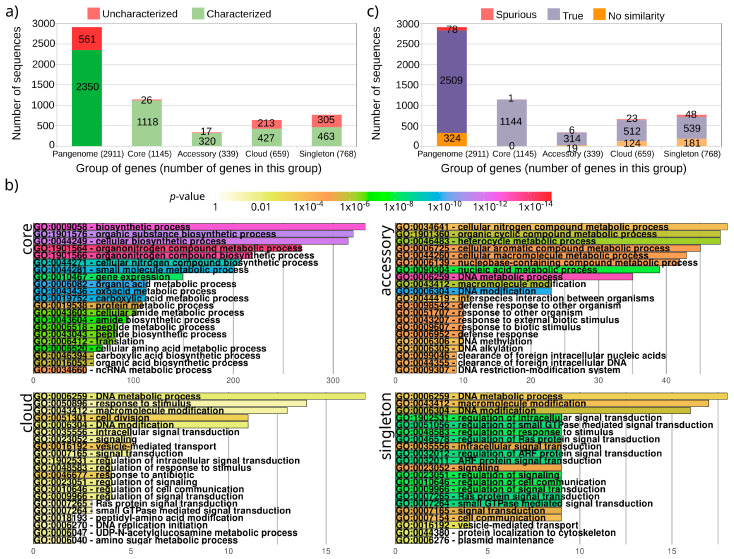
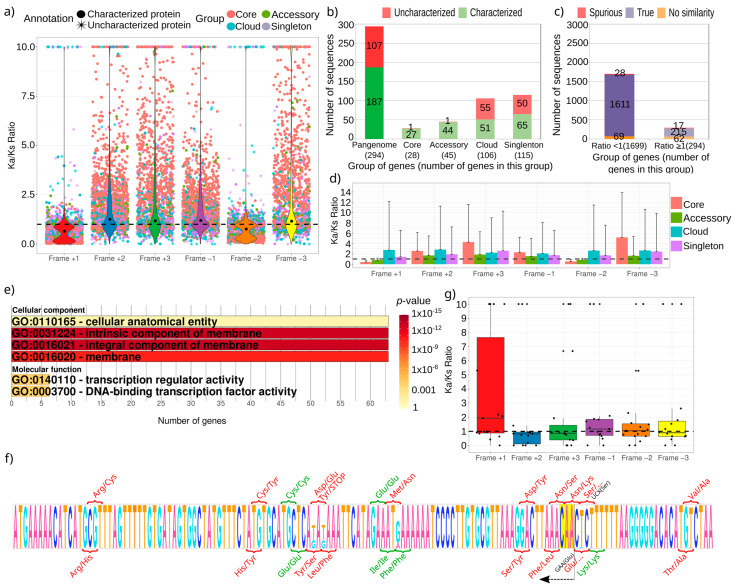
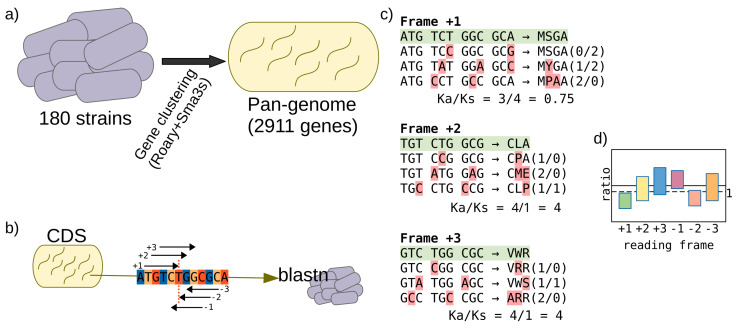
The current availability of complete genome sequences has allowed knowing that bacterial genomes can bear genes not present in the genome of all the strains from a specific species. So, the genes shared by all the strains comprise the core of the species, but the pangenome can be much greater and usually includes genes appearing in one only strain. Once the pangenome of a species is estimated, other studies can be undertaken to generate new knowledge, such as the study of the evolutionary selection for protein-coding genes. Most of the genes of a pangenome are expected to be subject to purifying selection that assures the conservation of function, especially those in the core group. However, some genes can be subject to selection pressure, such as genes involved in virulence that need to escape to the host immune system, which is more common in the accessory group of the pangenome. We analyzed 180 strains of , a bacterium that colonizes the gastric mucosa of half the world population and presents a low number of genes (around 1500 in a strain and 3000 in the pangenome). After the estimation of the pangenome, the evolutionary selection for each gene has been calculated, and we found that 85% of them are subject to purifying selection and the remaining genes present some grade of selection pressure. As expected, the latter group is enriched with genes encoding for membrane proteins putatively involved in interaction to host tissues. In addition, this group also presents a high number of uncharacterized genes and genes encoding for putative spurious proteins. It suggests that they could be false positives from the gene finders used for identifying them. All these results propose that this kind of analyses can be useful to validate gene predictions and functionally characterize proteins in complete genomes.
目前,完整基因组序列的可用性使得人们可以了解到,细菌基因组可能带有特定物种所有菌株基因组中不存在的基因。因此,所有菌株共有的基因构成了物种的核心,但泛基因组可能更大,通常包括仅在一个菌株中出现的基因。一旦估计了一个物种的泛基因组,就可以进行其他研究来生成新的知识,例如对蛋白质编码基因进化选择的研究。泛基因组的大多数基因预计会受到纯化选择的影响,以确保功能的保守性,尤其是核心组中的基因。然而,一些基因可能会受到选择压力的影响,例如参与毒力的基因需要逃避宿主免疫系统,这种情况在泛基因组的附加组中更为常见。我们分析了 180 株 ,这种细菌定植在世界上一半人口的胃黏膜上,其基因数量较少(一个菌株约有 1500 个基因,泛基因组中有 3000 个基因)。在估计了泛基因组之后,我们计算了每个基因的进化选择,发现其中 85%受到纯化选择的影响,其余基因则存在一定程度的选择压力。正如预期的那样,后者群体富含编码膜蛋白的基因,这些蛋白可能涉及与宿主组织的相互作用。此外,这个群体还存在大量未被表征的基因和编码假定假蛋白的基因。这表明它们可能是用于识别它们的基因发现者产生的假阳性。所有这些结果表明,这种分析可以用于验证基因预测,并对完整基因组中的蛋白质进行功能表征。