Oud Machteld M, Latour Brooke L, Bakey Zeineb, Letteboer Stef J, Lugtenberg Dorien, Wu Ka Man, Cornelissen Elisabeth A M, Yntema Helger G, Schmidts Miriam, Roepman Ronald, Bongers Ernie M H F
1Department of Human Genetics (855), Radboud University Medical Centre, PO-Box 9101, 6500 HB Nijmegen, The Netherlands.
2Radboud Institute for Molecular Life Sciences, Radboud University Medical Centre, PO-Box 9101, 6500 HB, Nijmegen, The Netherlands.
Cilia. 2018 Feb 23;7:1. doi: 10.1186/s13630-018-0055-2. eCollection 2018.
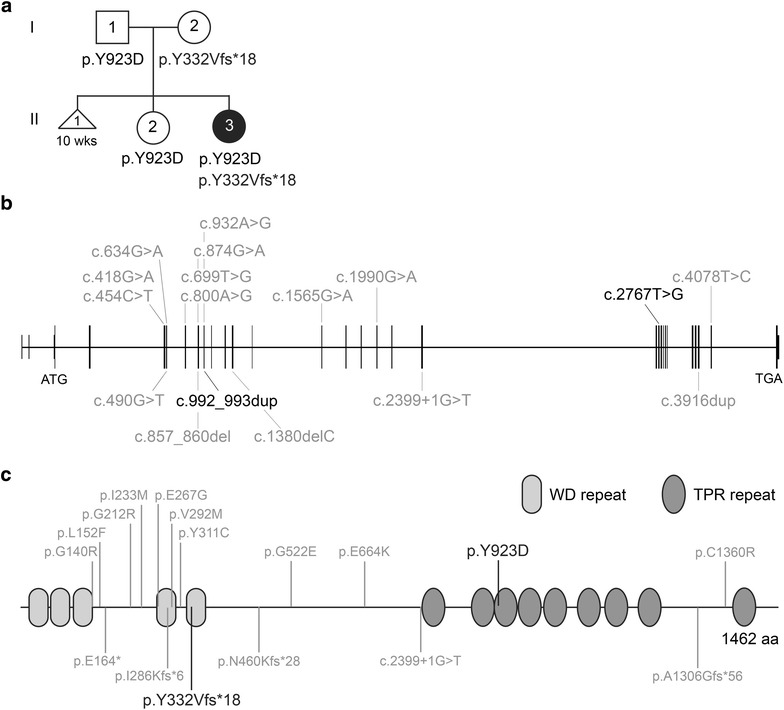
Mainzer-Saldino syndrome (MZSDS) is a skeletal ciliopathy and part of the short-rib thoracic dysplasia (SRTD) group of ciliary disorders. The main characteristics of MZSDS are short limbs, mild narrow thorax, blindness, and renal failure. Thus far, variants in two genes are associated with MZSDS: and . In this study, we describe a 1-year-old girl presenting with mild skeletal abnormalities, Leber congenital amaurosis, and bilateral hearing difficulties. For establishing an accurate diagnosis, we combined clinical, molecular, and functional analyses.
We performed diagnostic whole-exome sequencing (WES) analysis to determine the genetic cause of the disease and analyzed two gene panels, containing all currently known genes in vision disorders, and in hearing impairment. Upon detection of the likely causative variants, ciliary phenotyping was performed in patient urine-derived renal epithelial cells (URECs) and rescue experiments were performed in CRISPR/Cas9-derived knock out cells to determine the pathogenicity of the detected variants in vitro. Cilium morphology, cilium length, and intraflagellar transport (IFT) were evaluated by immunocytochemistry.
Diagnostic WES revealed two novel compound heterozygous variants in , encoding IFT140. Thorough investigation of WES data did not reveal any variants in candidate genes associated with hearing impairment. Patient-derived URECs revealed an accumulation of IFT-B protein IFT88 at the ciliary tip in 41% of the cells indicative of impaired retrograde IFT, while this was absent in cilia from control URECs. Furthermore, transfection of CRISPR/Cas9-derived knock out cells with an IFT140 construct containing the patient mutation p.Tyr923Asp resulted in a significantly higher percentage of IFT88 tip accumulation than transfection with the wild-type IFT140 construct.
By combining the clinical, genetic, and functional data from this study, we could conclude that the patient has SRTD9, also called Mainzer-Saldino syndrome, caused by variants in . We suggest the possibility that variants in may underlie hearing impairment. Moreover, we show that urine provides an excellent source to obtain patient-derived cells in a non-invasive manner to study the pathogenicity of variants detected by genetic testing.
梅恩泽尔-萨尔迪诺综合征(MZSDS)是一种骨骼纤毛病,属于纤毛疾病的短肋胸廓发育不良(SRTD)组。MZSDS的主要特征是四肢短小、胸廓轻度狭窄、失明和肾衰竭。迄今为止,两个基因的变异与MZSDS相关: 和 。在本研究中,我们描述了一名1岁女童,表现为轻度骨骼异常、莱伯先天性黑蒙和双侧听力障碍。为了建立准确的诊断,我们结合了临床、分子和功能分析。
我们进行了诊断性全外显子测序(WES)分析以确定疾病的遗传原因,并分析了两个基因panel,其中包含视力障碍和听力障碍方面所有目前已知的基因。在检测到可能的致病变异后,在患者尿液来源的肾上皮细胞(UREC)中进行纤毛表型分析,并在CRISPR/Cas9衍生的 敲除细胞中进行拯救实验,以在体外确定检测到的变异的致病性。通过免疫细胞化学评估纤毛形态、纤毛长度和鞭毛内运输(IFT)。
诊断性WES揭示了 基因中的两个新的复合杂合变异,该基因编码IFT140。对WES数据的全面调查未发现与听力障碍相关的候选基因中的任何变异。患者来源的UREC显示,41%的细胞中IFT-B蛋白IFT88在纤毛尖端积累,表明逆向IFT受损,而对照UREC的纤毛中不存在这种情况。此外,用含有患者突变p.Tyr923Asp的IFT140构建体转染CRISPR/Cas9衍生的 敲除细胞,导致IFT88尖端积累的百分比显著高于用野生型IFT140构建体转染。
通过结合本研究的临床、遗传和功能数据,我们可以得出结论,该患者患有SRTD9,也称为梅恩泽尔-萨尔迪诺综合征,由 基因的变异引起。我们提出 基因变异可能是听力障碍基础的可能性。此外,我们表明尿液提供了一个极好的来源,可以以非侵入性方式获得患者来源的细胞,以研究基因检测中检测到的变异的致病性。