Department of Biomedical Data Sciences, Section Molecular Epidemiology, Leiden University Medical Center, Leiden, The Netherlands.
The Delft Bioinformatics Lab, Delft University of Technology, Delft, The Netherlands.
Ann Rheum Dis. 2019 Feb;78(2):270-277. doi: 10.1136/annrheumdis-2018-213882. Epub 2018 Dec 1.
To uncover the microRNA (miRNA) interactome of the osteoarthritis (OA) pathophysiological process in the cartilage.
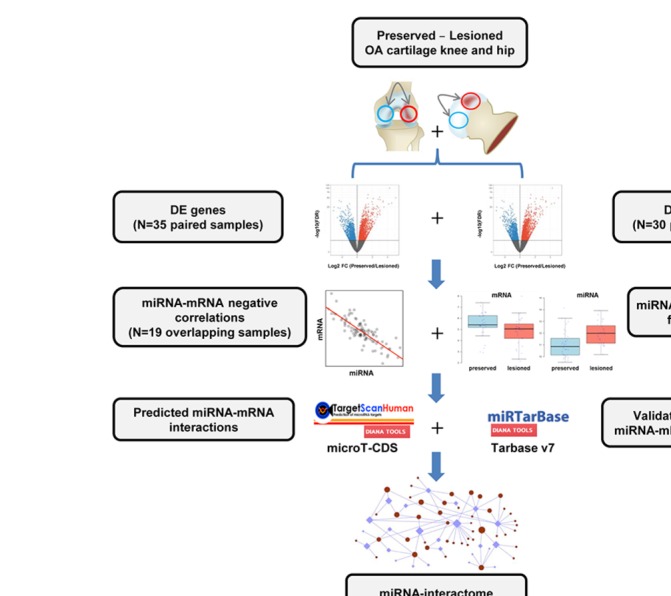
We performed RNA sequencing in 130 samples (n=35 and n=30 pairs for messenger RNA (mRNA) and miRNA, respectively) on macroscopically preserved and lesioned OA cartilage from the same patient and performed differential expression (DE) analysis of miRNA and mRNAs. To build an OA-specific miRNA interactome, a prioritisation scheme was applied based on inverse Pearson's correlations and inverse DE of miRNAs and mRNAs. Subsequently, these were filtered by those present in predicted (TargetScan/microT-CDS) and/or experimentally validated (miRTarBase/TarBase) public databases. Pathway enrichment analysis was applied to elucidate OA-related pathways likely mediated by miRNA regulatory mechanisms.
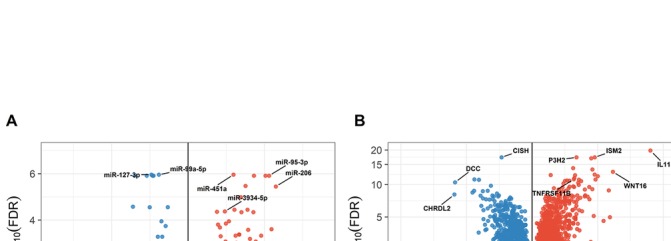
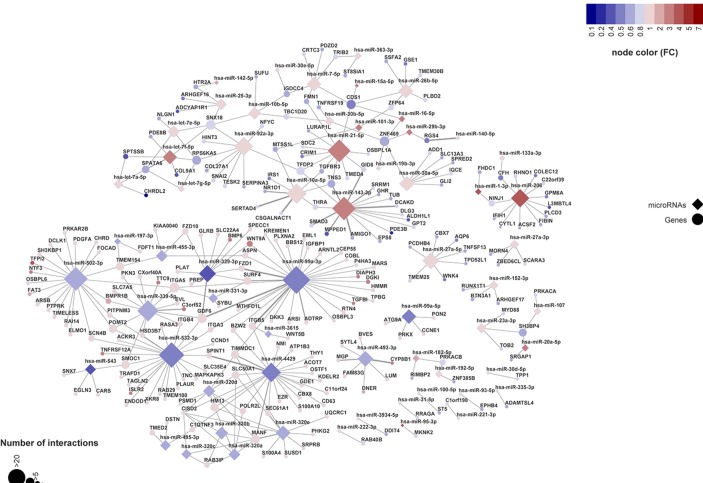
We found 142 miRNAs and 2387 mRNAs to be differentially expressed between lesioned and preserved OA articular cartilage. After applying prioritisation towards likely miRNA-mRNA targets, a regulatory network of 62 miRNAs targeting 238 mRNAs was created. Subsequent pathway enrichment analysis of these mRNAs (or genes) elucidated that genes within the 'nervous system development' are likely mediated by miRNA regulatory mechanisms (familywise error=8.4×10). Herein encodes neurotrophin-3, which controls survival and differentiation of neurons and which is closely related to the nerve growth factor.
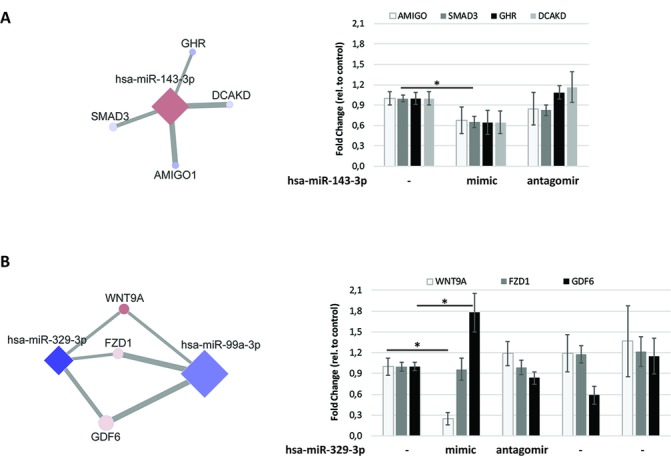
By an integrated approach of miRNA and mRNA sequencing data of OA cartilage, an OA miRNA interactome and related pathways were elucidated. Our functional data demonstrated interacting levels at which miRNA affects expression of genes in the cartilage and exemplified the complexity of functionally validating a network of genes that may be targeted by multiple miRNAs.
揭示软骨骨关节炎(OA)病理过程中的 microRNA(miRNA)互作组。
我们对 130 个样本(mRNA 为 n=35 对,miRNA 为 n=30 对)进行了 RNA 测序,这些样本来自同一患者的宏观保存和病变 OA 软骨,并对 miRNA 和 mRNAs 进行了差异表达(DE)分析。为构建特定于 OA 的 miRNA 互作组,我们应用了一种基于 miRNA 和 mRNAs 的逆 Pearson 相关性和逆 DE 的优先级方案。随后,通过预测(TargetScan/microT-CDS)和/或实验验证(miRTarBase/TarBase)公共数据库中存在的 miRNA 进行过滤。应用通路富集分析阐明可能由 miRNA 调节机制介导的 OA 相关通路。
我们发现病变和保存的 OA 关节软骨之间有 142 个 miRNA 和 2387 个 mRNAs 存在差异表达。在对可能的 miRNA-mRNA 靶标进行优先级排序后,创建了一个由 62 个 miRNA 靶向 238 个 mRNAs 的调控网络。随后对这些 mRNAs(或基因)进行通路富集分析表明,“神经系统发育”中的基因可能受 miRNA 调节机制调控(家族错误率=8.4×10)。编码神经营养因子-3,它控制神经元的存活和分化,与神经生长因子密切相关。
通过 OA 软骨 miRNA 和 mRNA 测序数据的综合方法,阐明了 OA miRNA 互作组及其相关通路。我们的功能数据表明,miRNA 以交互的方式影响软骨中基因的表达,并举例说明了验证可能受多个 miRNA 靶向的基因网络的复杂性。