MRC Centre for Outbreak Analysis and Modelling, Department of Infectious Disease Epidemiology, School of Public Health, Imperial College London, London, United Kingdom.
School of Life Sciences, University of Sussex, Brighton, United Kingdom.
PLoS Comput Biol. 2018 Dec 17;14(12):e1006554. doi: 10.1371/journal.pcbi.1006554. eCollection 2018 Dec.
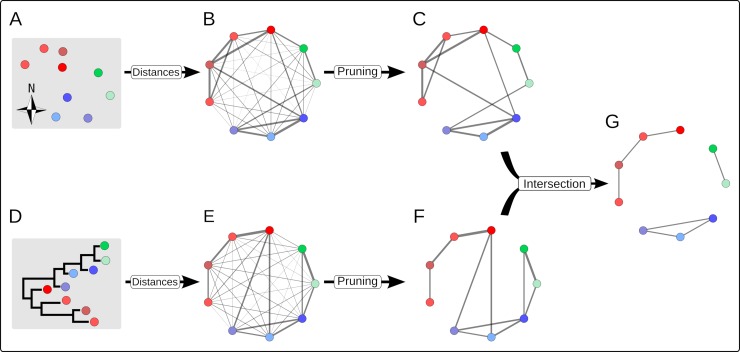
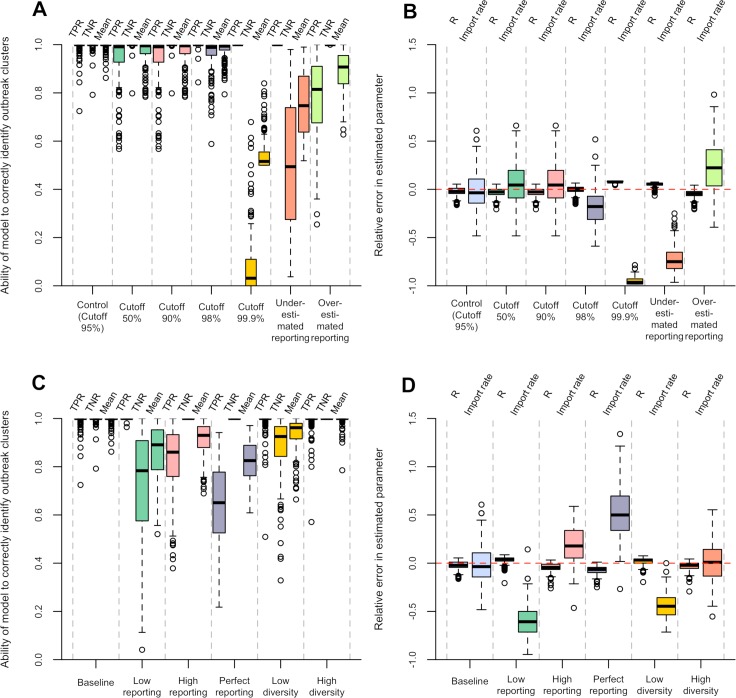
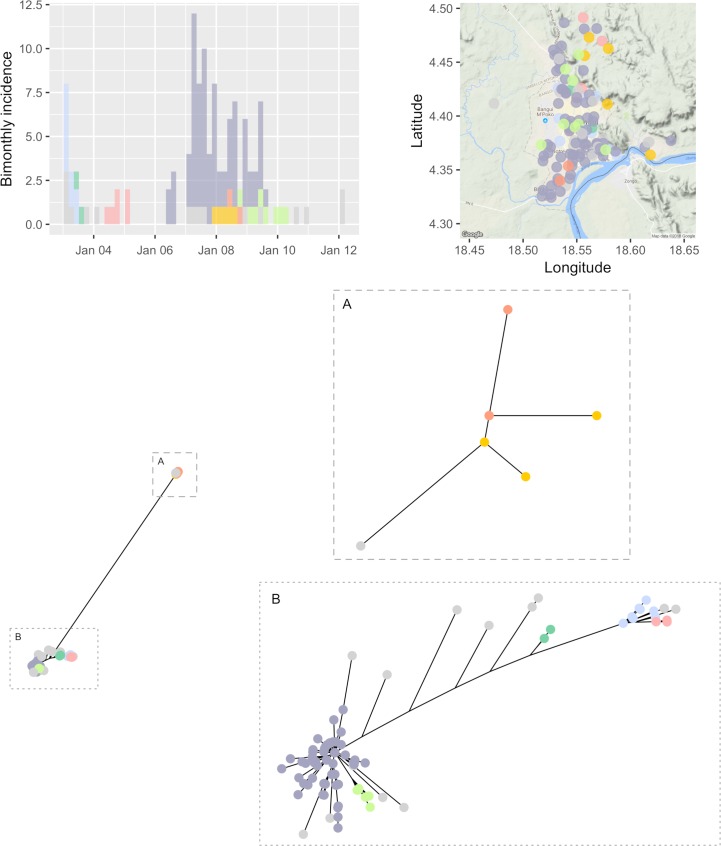
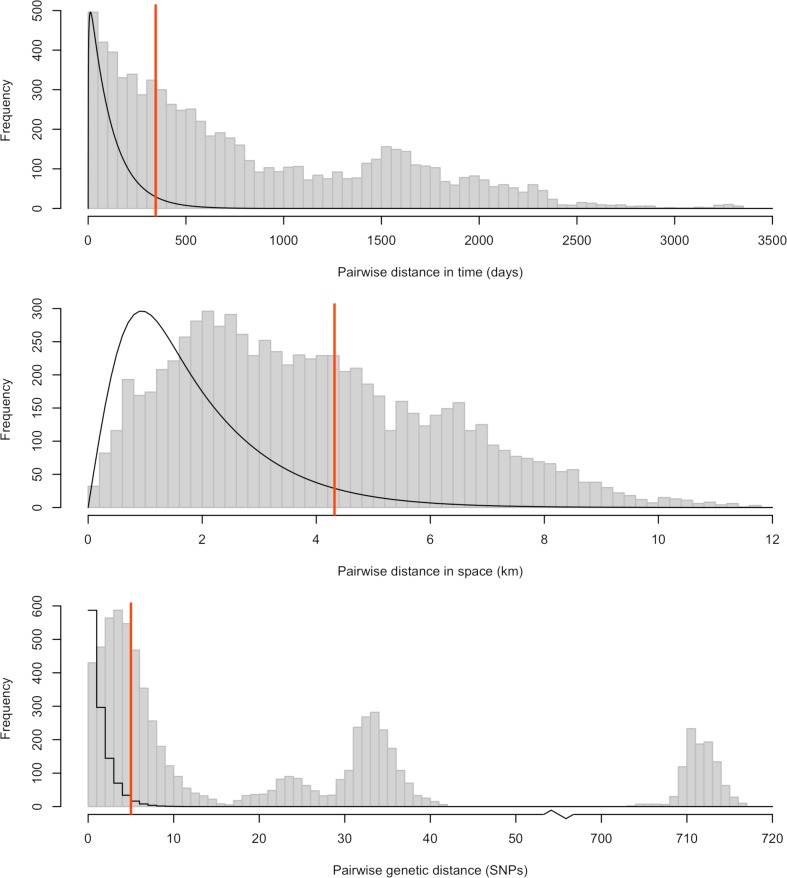
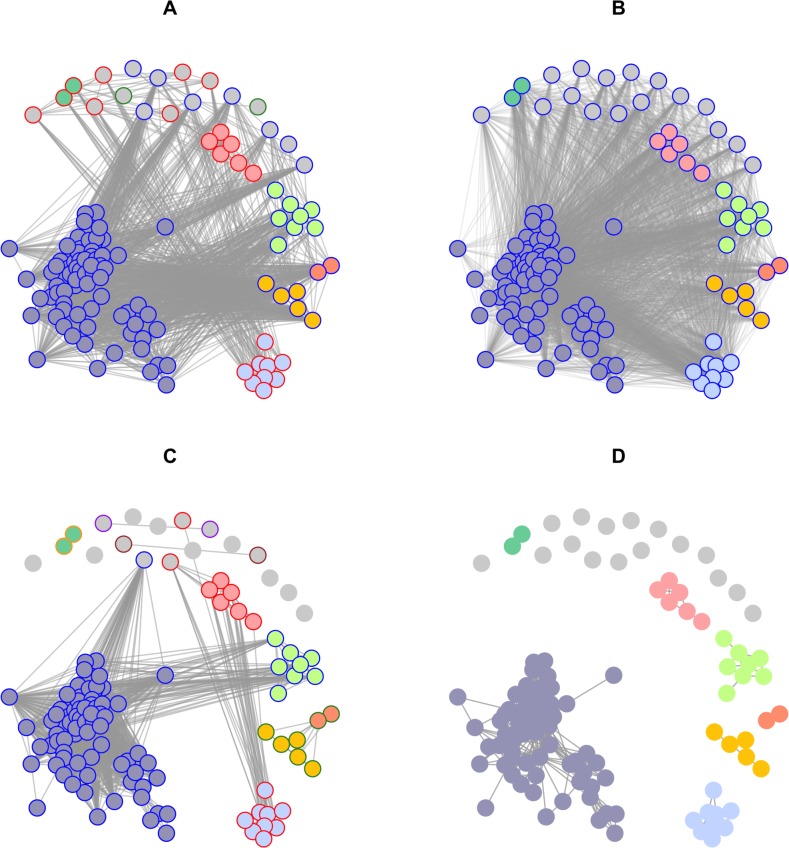
Early assessment of infectious disease outbreaks is key to implementing timely and effective control measures. In particular, rapidly recognising whether infected individuals stem from a single outbreak sustained by local transmission, or from repeated introductions, is crucial to adopt effective interventions. In this study, we introduce a new framework for combining several data streams, e.g. temporal, spatial and genetic data, to identify clusters of related cases of an infectious disease. Our method explicitly accounts for underreporting, and allows incorporating preexisting information about the disease, such as its serial interval, spatial kernel, and mutation rate. We define, for each data stream, a graph connecting all cases, with edges weighted by the corresponding pairwise distance between cases. Each graph is then pruned by removing distances greater than a given cutoff, defined based on preexisting information on the disease and assumptions on the reporting rate. The pruned graphs corresponding to different data streams are then merged by intersection to combine all data types; connected components define clusters of cases related for all types of data. Estimates of the reproduction number (the average number of secondary cases infected by an infectious individual in a large population), and the rate of importation of the disease into the population, are also derived. We test our approach on simulated data and illustrate it using data on dog rabies in Central African Republic. We show that the outbreak clusters identified using our method are consistent with structures previously identified by more complex, computationally intensive approaches.
传染病暴发的早期评估是实施及时有效的控制措施的关键。特别是,迅速识别受感染个体是源自本地传播的单一暴发,还是源自反复传入,对于采取有效的干预措施至关重要。在本研究中,我们引入了一种新的框架,用于结合多个数据流,例如时间、空间和遗传数据,以识别传染病相关病例的集群。我们的方法明确考虑了漏报情况,并允许纳入有关疾病的现有信息,例如其传播间隔、空间核和突变率。我们为每个数据流定义了一个连接所有病例的图,边的权重由病例之间的相应成对距离决定。然后,根据疾病的现有信息和报告率的假设,通过删除大于给定截止值的距离来修剪每个图。来自不同数据流的修剪图通过交集合并,以组合所有数据类型;连接组件定义了所有数据类型的相关病例集群。还得出了繁殖数(在大人群中,一个传染病个体感染的继发病例的平均数量)和疾病传入人口的速率的估计值。我们在模拟数据上测试了我们的方法,并使用中非共和国狗狂犬病的数据进行了说明。我们表明,使用我们的方法识别的暴发集群与先前通过更复杂、计算密集型方法识别的结构一致。