Guzmán Gabriela I, Olson Connor A, Hefner Ying, Phaneuf Patrick V, Catoiu Edward, Crepaldi Lais B, Micas Lucas Goldschmidt, Palsson Bernhard O, Feist Adam M
Department of Bioengineering, University of California, San Diego, La Jolla, 92093, CA, USA.
Department of Bioinformatics and Systems Biology, University of California, San Diego, 92093, La Jolla, CA, USA.
BMC Syst Biol. 2018 Dec 17;12(1):143. doi: 10.1186/s12918-018-0653-z.
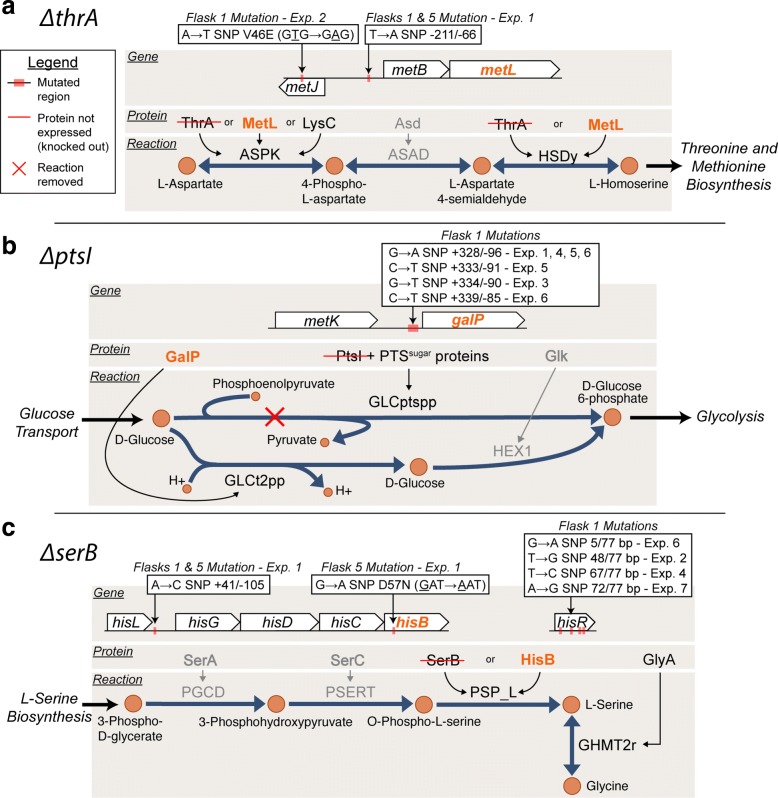
Essentiality assays are important tools commonly utilized for the discovery of gene functions. Growth/no growth screens of single gene knockout strain collections are also often utilized to test the predictive power of genome-scale models. False positive predictions occur when computational analysis predicts a gene to be non-essential, however experimental screens deem the gene to be essential. One explanation for this inconsistency is that the model contains the wrong information, possibly an incorrectly annotated alternative pathway or isozyme reaction. Inconsistencies could also be attributed to experimental limitations, such as growth tests with arbitrary time cut-offs. The focus of this study was to resolve such inconsistencies to better understand isozyme activities and gene essentiality.
In this study, we explored the definition of conditional essentiality from a phenotypic and genomic perspective. Gene-deletion strains associated with false positive predictions of gene essentiality on defined minimal medium for Escherichia coli were targeted for extended growth tests followed by population sequencing and transcriptome analysis. Of the twenty false positive strains available and confirmed from the Keio single gene knock-out collection, 11 strains were shown to grow with longer incubation periods making these actual true positives. These strains grew reproducibly with a diverse range of growth phenotypes. The lag phase observed for these strains ranged from less than one day to more than 7 days. It was found that 9 out of 11 of the false positive strains that grew acquired mutations in at least one replicate experiment and the types of mutations ranged from SNPs and small indels associated with regulatory or metabolic elements to large regions of genome duplication. Comparison of the detected adaptive mutations, modeling predictions of alternate pathways and isozymes, and transcriptome analysis of KO strains suggested agreement for the observed growth phenotype for 6 out of the 9 cases where mutations were observed.
Longer-term growth experiments followed by whole genome sequencing and transcriptome analysis can provide a better understanding of conditional gene essentiality and mechanisms of adaptation to such perturbations. Compensatory mutations are largely reproducible mechanisms and are in agreement with genome-scale modeling predictions to loss of function gene deletion events.
必需性分析是常用于发现基因功能的重要工具。单基因敲除菌株文库的生长/非生长筛选也经常用于测试基因组规模模型的预测能力。当计算分析预测一个基因是非必需的,但实验筛选却认为该基因是必需的时,就会出现假阳性预测。这种不一致的一种解释是模型包含错误信息,可能是注释错误的替代途径或同工酶反应。不一致也可能归因于实验限制,例如具有任意时间截止点的生长测试。本研究的重点是解决此类不一致问题,以更好地理解同工酶活性和基因必需性。
在本研究中,我们从表型和基因组角度探讨了条件必需性的定义。针对在大肠杆菌的限定基本培养基上基因必需性的假阳性预测相关的基因缺失菌株进行延长生长测试,随后进行群体测序和转录组分析。在从Keio单基因敲除文库中获得并确认的20个假阳性菌株中,有11个菌株在延长培养期后显示出能够生长,这表明它们实际上是真阳性。这些菌株以多种生长表型可重复生长。观察到这些菌株的滞后期从不到一天到超过7天不等。发现11个生长的假阳性菌株中有9个在至少一次重复实验中获得了突变,突变类型从与调控或代谢元件相关的单核苷酸多态性(SNP)和小插入缺失到基因组大片段重复。对检测到的适应性突变、替代途径和同工酶的建模预测以及敲除菌株的转录组分析表明,在观察到突变的9个案例中有6个案例中观察到的生长表型是一致的。
随后进行全基因组测序和转录组分析的长期生长实验可以更好地理解条件基因必需性以及适应此类扰动的机制。补偿性突变在很大程度上是可重复的机制,并且与基因组规模模型对功能丧失基因缺失事件的预测一致。