Stanford Cardiovascular Institute (T.S., R.S., C.K.L., C.C., W.L.M., E.L., A.W., M.G, J.L., A.O., S.L., H.Y., M.M., M.W., E.A.A., I.K., J.C.W.), Stanford University School of Medicine, CA.
Department of Medicine, Division of Cardiology (T.S., R.S., C.K.L., C.C., W.L.M., E.L., J.L., A.O., S.L., H.Y., M.M., M.W., E.A.A., J.C.W.), Stanford University School of Medicine, CA.
Circulation. 2019 Feb 5;139(6):799-811. doi: 10.1161/CIRCULATIONAHA.118.034624.
Hypertrophic cardiomyopathy (HCM) is frequently caused by mutations in myosin-binding protein C3 ( MYBPC3) resulting in a premature termination codon (PTC). The underlying mechanisms of how PTC mutations in MYBPC3 lead to the onset and progression of HCM are poorly understood. This study's aim was to investigate the molecular mechanisms underlying the pathogenesis of HCM associated with MYBPC3 PTC mutations by utilizing human isogenic induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs).
Isogenic iPSC lines were generated from HCM patients harboring MYBPC3 PTC mutations (p.R943x; p.R1073P_Fsx4) using genome editing. Comprehensive phenotypic and transcriptome analyses were performed in the iPSC-CMs.
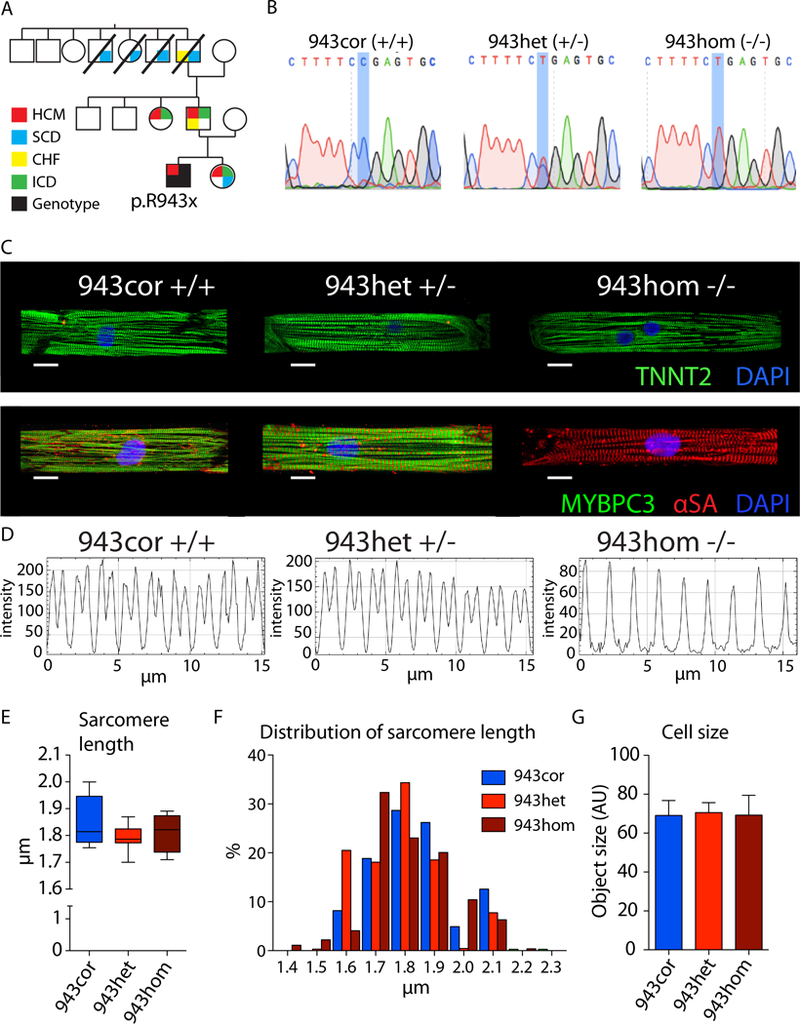
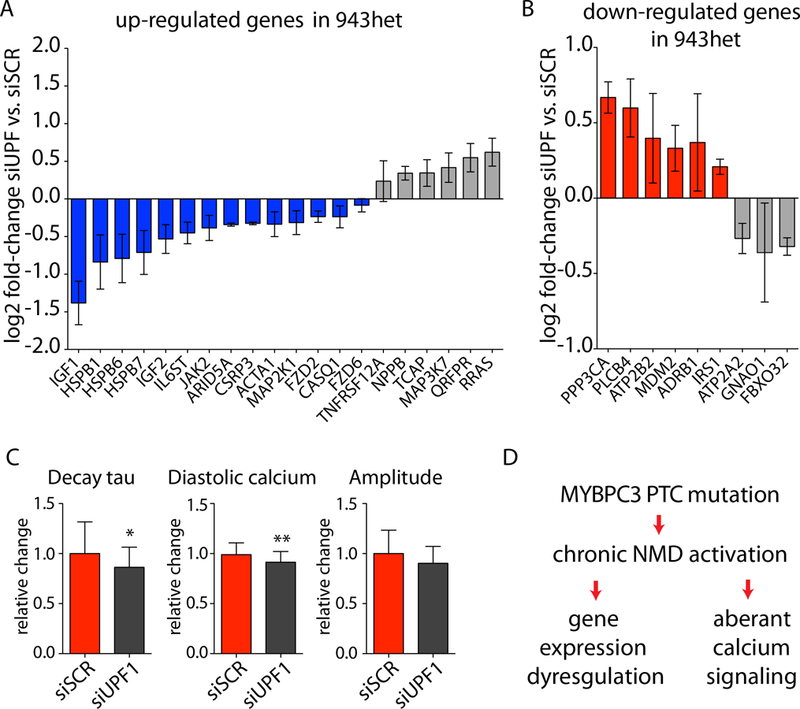
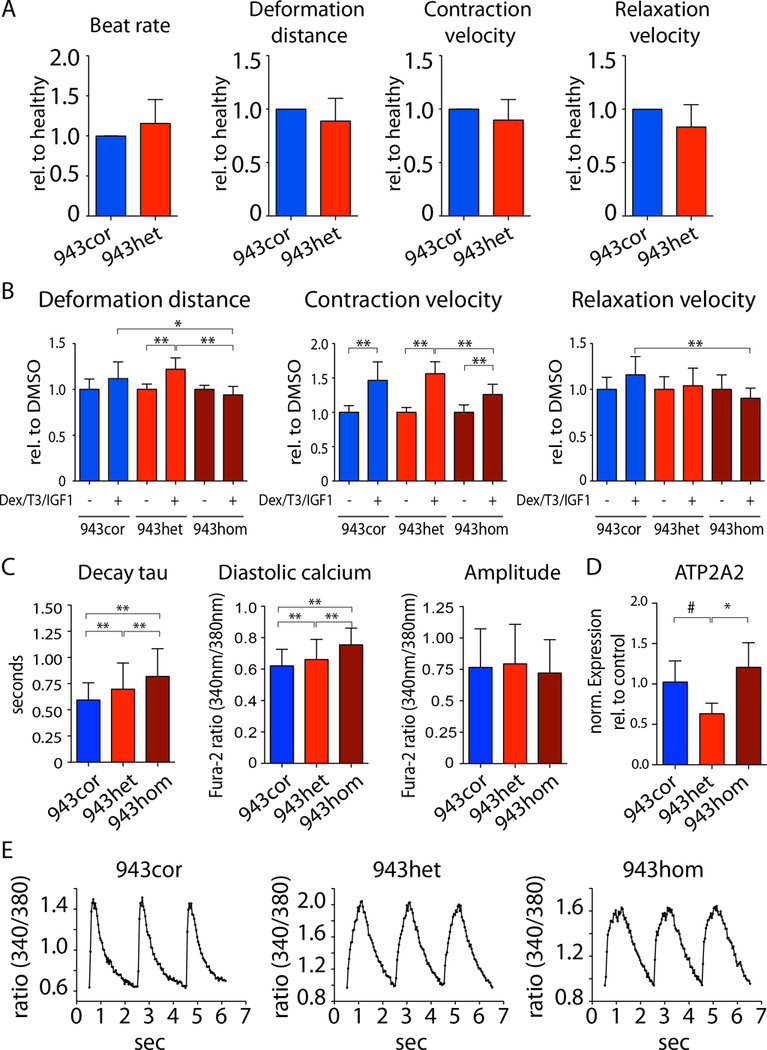
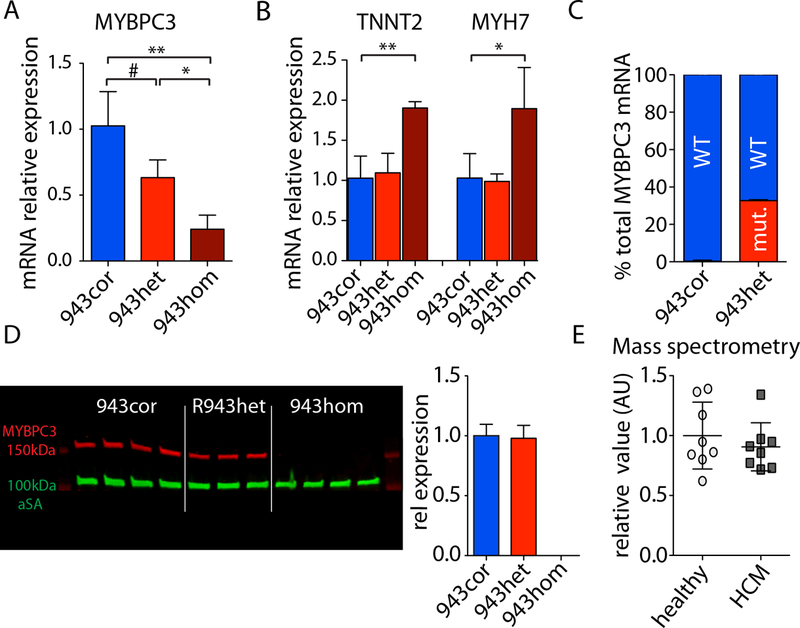
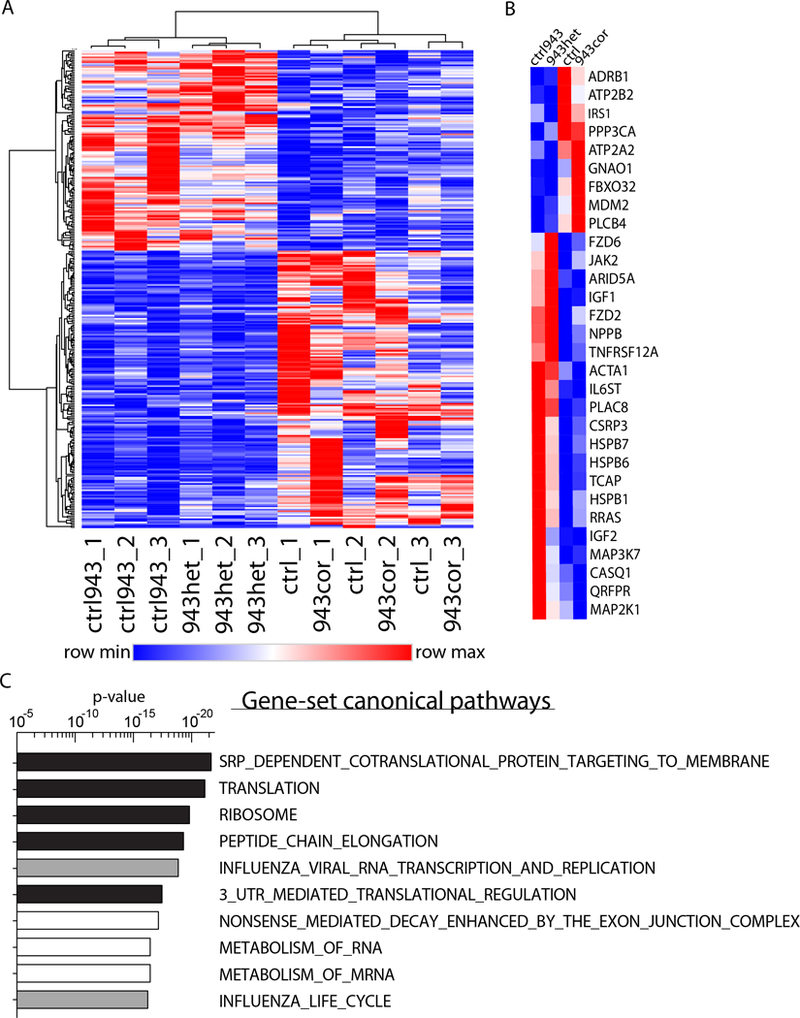
We observed aberrant calcium handling properties with prolonged decay kinetics and elevated diastolic calcium levels in the absence of structural abnormalities or contracile dysfunction in HCM iPSC-CMs as compared to isogenic controls. The mRNA expression levels of MYBPC3 were significantly reduced in mutant iPSC-CMs, but the protein levels were comparable among isogenic iPSC-CMs, suggesting that haploinsufficiency of MYBPC3 does not contribute to the pathogenesis of HCM in vitro. Furthermore, truncated MYBPC3 peptides were not detected. At the molecular level, the nonsense-mediated decay pathway was activated, and a set of genes involved in major cardiac signaling pathways was dysregulated in HCM iPSC-CMs, indicating an HCM gene signature in vitro. Specific inhibition of the nonsense-mediated decay pathway in mutant iPSC-CMs resulted in reversal of the molecular phenotype and normalization of calcium-handling abnormalities.
iPSC-CMs carrying MYBPC3 PTC mutations displayed aberrant calcium signaling and molecular dysregulations in the absence of significant haploinsufficiency of MYBPC3 protein. Here we provided the first evidence of the direct connection between the chronically activated nonsense-mediated decay pathway and HCM disease development.
肥厚型心肌病(HCM)常由肌球蛋白结合蛋白 C3(MYBPC3)的突变引起,导致提前终止密码子(PTC)。目前对于 MYBPC3 中的 PTC 突变如何导致 HCM 的发生和进展的潜在机制还了解甚少。本研究旨在利用人同基因诱导多能干细胞衍生的心肌细胞(iPSC-CMs)来研究与 MYBPC3 PTC 突变相关的 HCM 发病机制的分子机制。
使用基因组编辑从携带 MYBPC3 PTC 突变(p.R943x;p.R1073P_Fsx4)的 HCM 患者中生成同基因 iPSC 系。在 iPSC-CMs 中进行全面的表型和转录组分析。
与同基因对照相比,在 absence of 结构异常或收缩功能障碍的情况下,HCM iPSC-CMs 表现出异常的钙处理特性,钙衰减动力学延长,舒张钙水平升高。突变型 iPSC-CMs 中的 MYBPC3 mRNA 表达水平显著降低,但同基因 iPSC-CMs 中的蛋白水平相当,提示 MYBPC3 的杂合不足在体外不会导致 HCM 的发病机制。此外,未检测到截断的 MYBPC3 肽。在分子水平上,无意义介导的衰变途径被激活,并且在 HCM iPSC-CMs 中一组参与主要心脏信号通路的基因失调,表明体外存在 HCM 基因特征。在突变型 iPSC-CMs 中特异性抑制无意义介导的衰变途径可逆转分子表型并使钙处理异常正常化。
携带 MYBPC3 PTC 突变的 iPSC-CMs 在 MYBPC3 蛋白明显杂合不足的情况下表现出异常的钙信号和分子失调。在这里,我们首次提供了慢性激活的无意义介导的衰变途径与 HCM 疾病发展之间直接联系的证据。