Suthiworachai Chanisara, Tammachote Rachaneekorn, Srichomthong Chalurmpon, Ittiwut Rungnapa, Suphapeetiporn Kanya, Sahakitrungruang Taninee, Shotelersuk Vorasuk
Biological Sciences Program, Faculty of Science, Chulalongkorn University, Bangkok, Thailand.
Department of Botany, Faculty of Science, Chulalongkorn University, Bangkok, Thailand.
J Endocr Soc. 2018 Dec 12;3(1):171-180. doi: 10.1210/js.2018-00270. eCollection 2019 Jan 1.
() mutations cause X-linked adrenal hypoplasia congenita (AHC) and hypogonadotropic hypogonadism (HH) in affected male patients. Affected individuals typically present with early-onset adrenal insufficiency and develop HH during puberty. Rare cases can present with late-onset adrenal insufficiency or other unusual phenotypes.
We sought to identify and functionally characterize mutations in seven Thai male subjects in six families with X-linked AHC.
Six patients had classic phenotypes with early-onset adrenal failure. One patient presented with late-onset Addison disease at 17 years. In the early-onset group, one patient had GnRH-independent sexual precocity at 3 years of age, and another patient had growth hormone deficiency. The gene was sequenced from all patients, and the transcriptional activities of the identified mutations were assessed using luciferase assays.
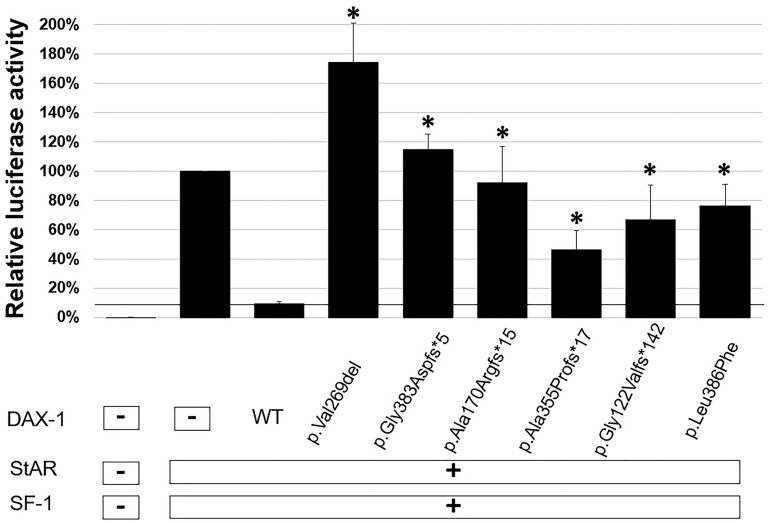
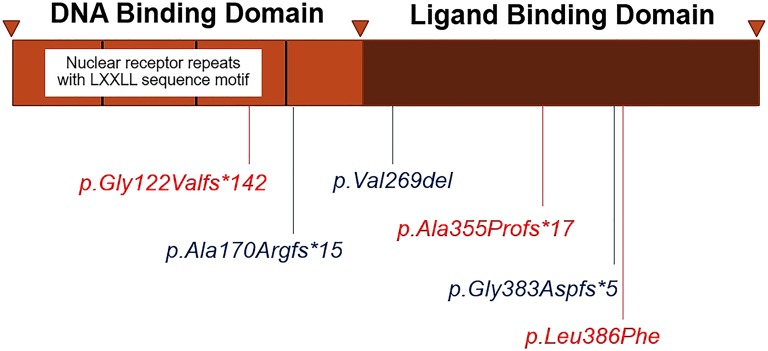
mutations were identified in all patients, including three novel mutations [c.363delG (p.Gly122Valfs142), c.1062delC (p.Ala355Profs17), and c.1156C>T (p.Leu386Phe)] and three known mutations [c.1148_1149delGG (p.Gly383Aspfs5), c.501_502insG (p.Ala170Argfs15), and c.805_807delGTC (p.Val269del)]. Functional studies showed that the mutants had lower levels of repressor activity on the gene promoter compared with the wild-type DAX-1 protein.
This study describes unusual phenotypes and three novel mutations, extending the phenotypic and mutational spectra of mutations.
()突变导致受影响男性患者出现X连锁先天性肾上腺发育不全(AHC)和低促性腺激素性性腺功能减退(HH)。受影响个体通常表现为早发性肾上腺功能不全,并在青春期发展为HH。罕见病例可表现为迟发性肾上腺功能不全或其他不寻常的表型。
我们试图在6个患有X连锁AHC的泰国家庭的7名男性受试者中鉴定并对()突变进行功能表征。
6例患者具有早发性肾上腺功能衰竭的典型表型。1例患者在17岁时出现迟发性Addison病。在早发性组中,1例患者在3岁时出现GnRH非依赖性性早熟,另1例患者有生长激素缺乏。对所有患者的()基因进行测序,并使用荧光素酶测定法评估所鉴定突变的转录活性。
在所有患者中均鉴定到()突变,包括3个新突变[c.363delG(p.Gly122Valfs142)、c.1062delC(p.Ala355Profs17)和c.1156C>T(p.Leu386Phe)]和3个已知突变[c.1148_1149delGG(p.Gly383Aspfs5)、c.501_502insG(p.Ala170Argfs15)和c.805_807delGTC(p.Val269del)]。功能研究表明,与野生型DAX-1蛋白相比,()突变体对()基因启动子的抑制活性水平较低。
本研究描述了不寻常的表型和3个新突变,扩展了()突变的表型和突变谱。