Institute of Integrative Biology, University of Liverpool, Liverpool, United Kingdom.
Department of Microbiology and Immunology, Stanford University School of Medicine, Stanford, California, United States of America.
PLoS Biol. 2019 Jan 15;17(1):e3000059. doi: 10.1371/journal.pbio.3000059. eCollection 2019 Jan.
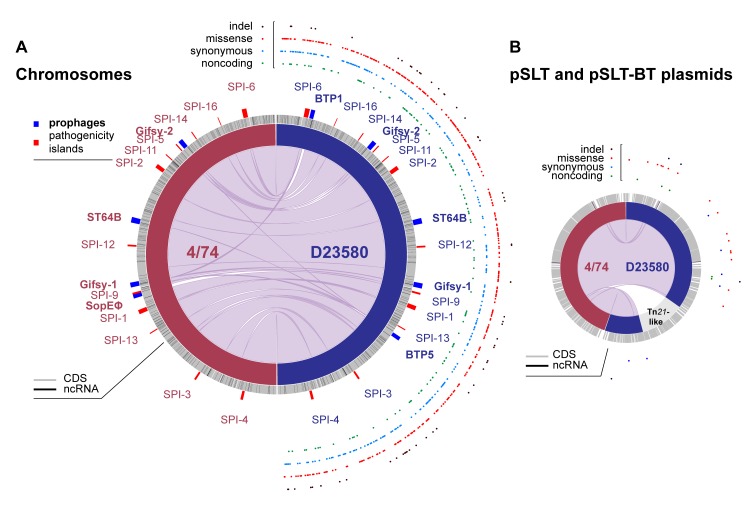
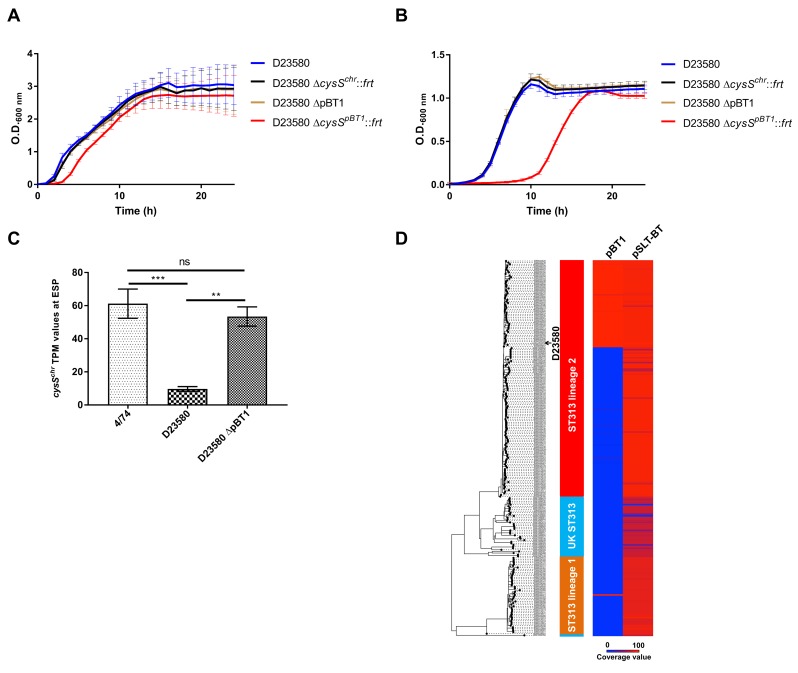
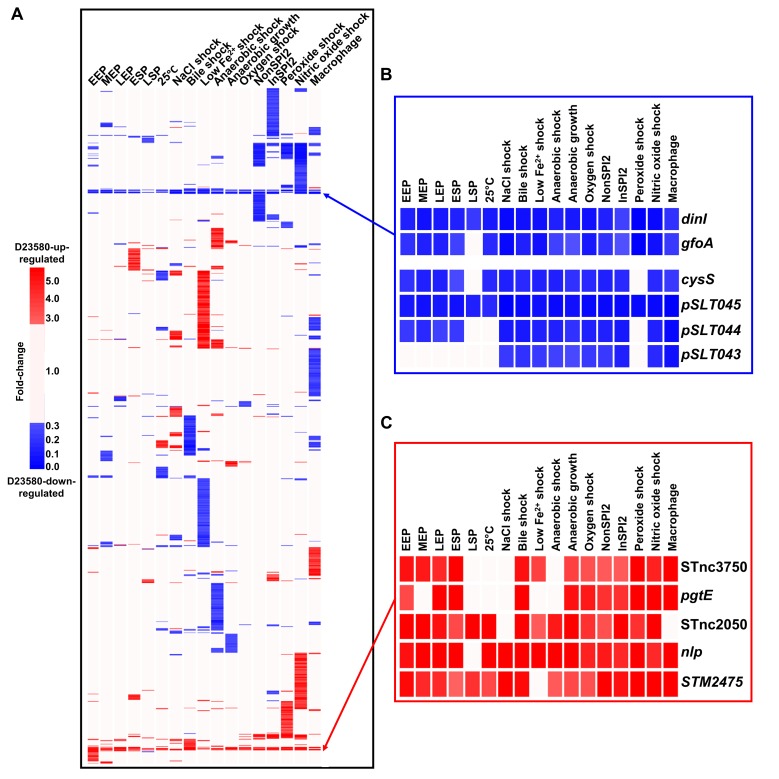
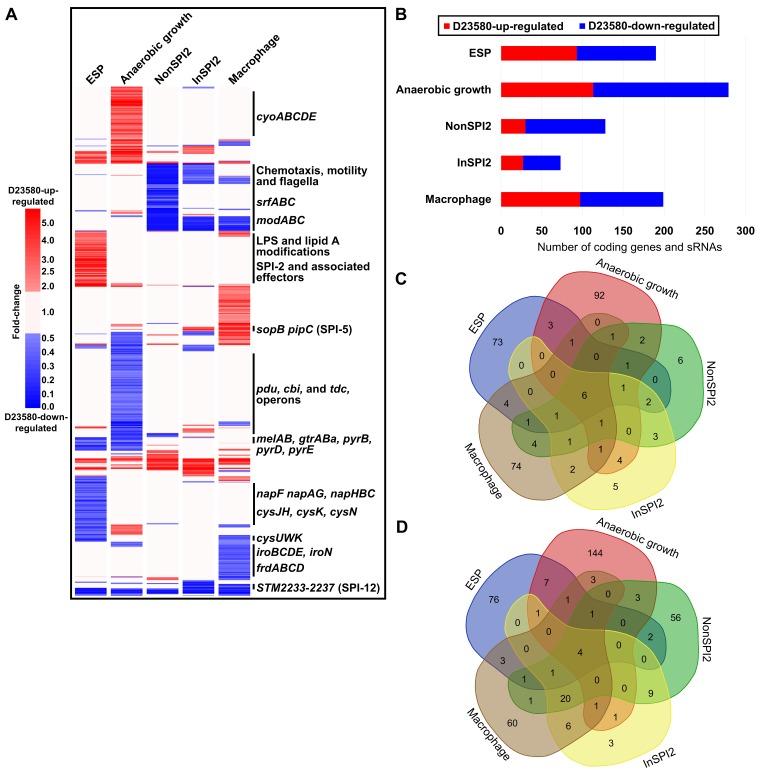
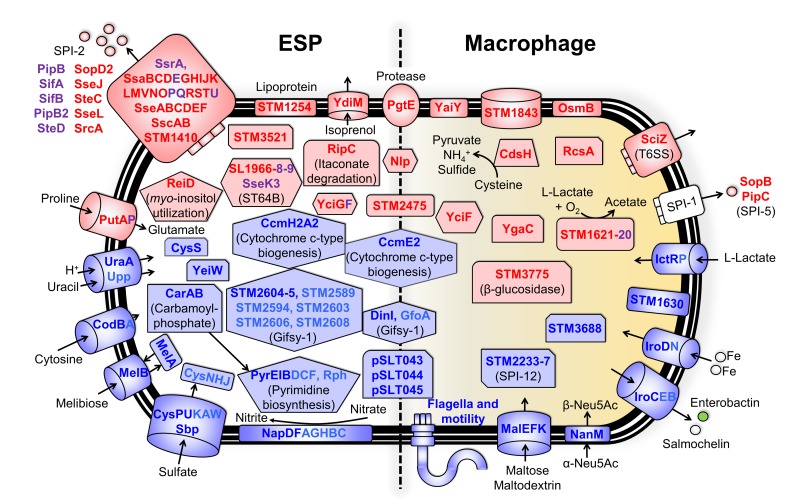
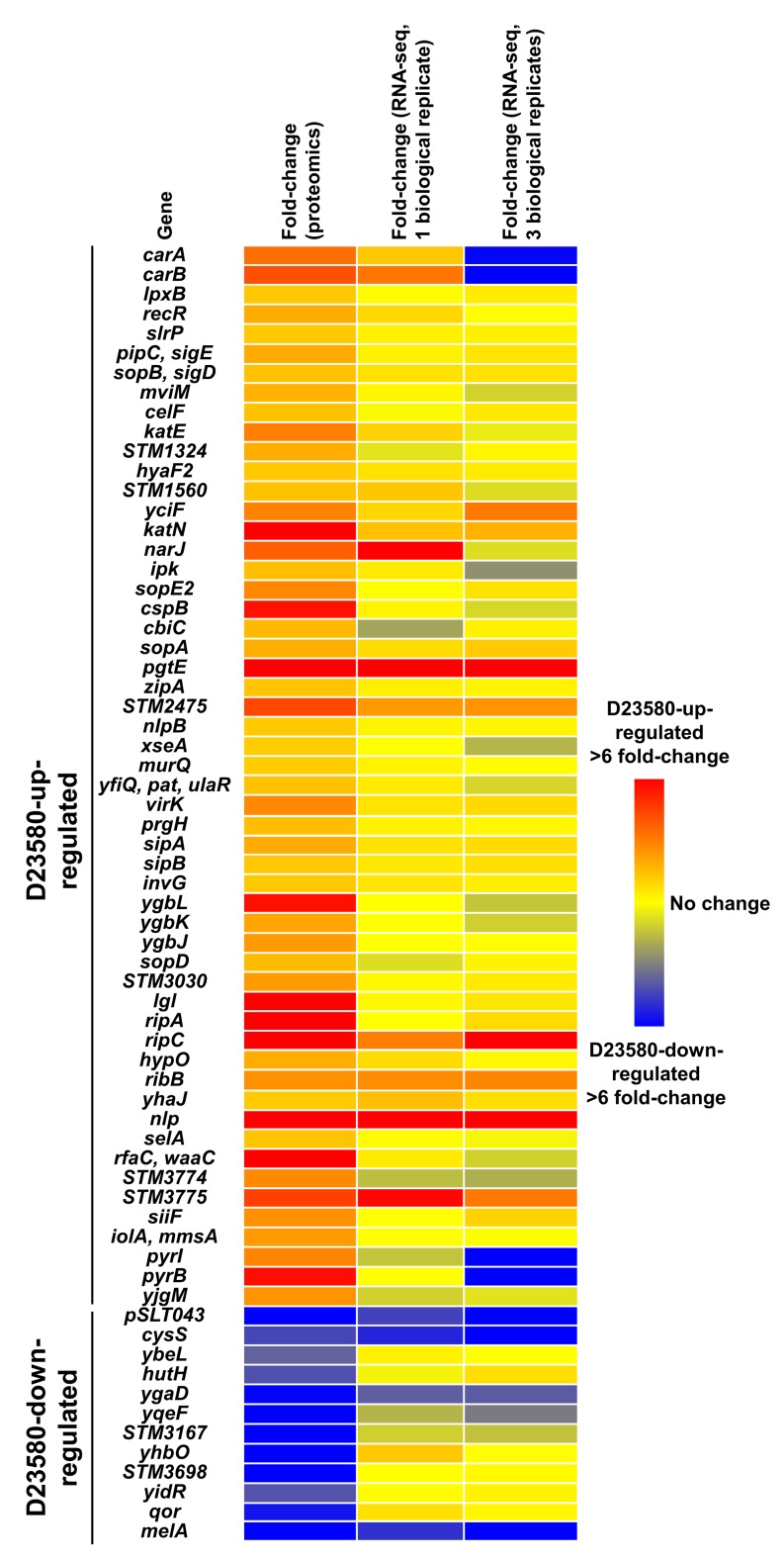
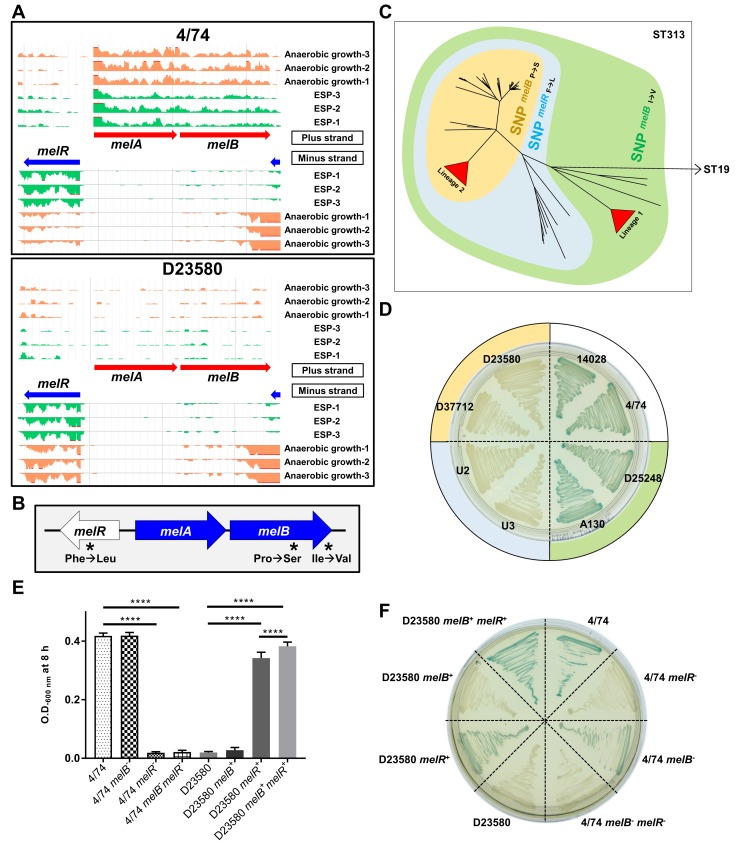
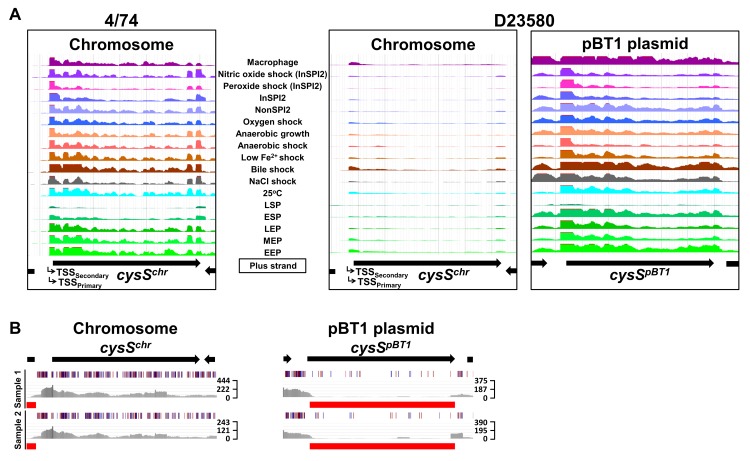
Salmonella Typhimurium sequence type (ST) 313 causes invasive nontyphoidal Salmonella (iNTS) disease in sub-Saharan Africa, targeting susceptible HIV+, malarial, or malnourished individuals. An in-depth genomic comparison between the ST313 isolate D23580 and the well-characterized ST19 isolate 4/74 that causes gastroenteritis across the globe revealed extensive synteny. To understand how the 856 nucleotide variations generated phenotypic differences, we devised a large-scale experimental approach that involved the global gene expression analysis of strains D23580 and 4/74 grown in 16 infection-relevant growth conditions. Comparison of transcriptional patterns identified virulence and metabolic genes that were differentially expressed between D23580 versus 4/74, many of which were validated by proteomics. We also uncovered the S. Typhimurium D23580 and 4/74 genes that showed expression differences during infection of murine macrophages. Our comparative transcriptomic data are presented in a new enhanced version of the Salmonella expression compendium, SalComD23580: http://bioinf.gen.tcd.ie/cgi-bin/salcom_v2.pl. We discovered that the ablation of melibiose utilization was caused by three independent SNP mutations in D23580 that are shared across ST313 lineage 2, suggesting that the ability to catabolize this carbon source has been negatively selected during ST313 evolution. The data revealed a novel, to our knowledge, plasmid maintenance system involving a plasmid-encoded CysS cysteinyl-tRNA synthetase, highlighting the power of large-scale comparative multicondition analyses to pinpoint key phenotypic differences between bacterial pathovariants.
肠炎沙门氏菌血清型 313(Salmonella Typhimurium sequence type 313,ST313)在撒哈拉以南非洲地区引起侵袭性非伤寒沙门氏菌(invasive nontyphoidal Salmonella,iNTS)病,主要针对易感的 HIV+、疟疾或营养不良个体。ST313 分离株 D23580 与全球范围内引起肠胃炎的高度特征化 ST19 分离株 4/74 之间的深入基因组比较显示出广泛的同线性。为了了解 856 个核苷酸变异如何产生表型差异,我们设计了一种大规模实验方法,该方法涉及在 16 种与感染相关的生长条件下,对菌株 D23580 和 4/74 的全局基因表达进行分析。转录模式的比较确定了 D23580 与 4/74 之间差异表达的毒力和代谢基因,其中许多基因通过蛋白质组学得到了验证。我们还发现了在鼠巨噬细胞感染期间表现出表达差异的鼠伤寒沙门氏菌 D23580 和 4/74 基因。我们的比较转录组数据在一个新的增强版沙门氏菌表达汇编 SalComD23580 中呈现,网址为:http://bioinf.gen.tcd.ie/cgi-bin/salcom_v2.pl。我们发现,在 D23580 中,有三个独立的单核苷酸多态性(SNP)突变导致了棉子糖利用的缺失,这三个 SNP 突变在 ST313 谱系 2 中是共有的,这表明在 ST313 进化过程中,这种碳源的代谢能力已被负选择。该数据揭示了一种新颖的、据我们所知的质粒维持系统,涉及一种质粒编码的半胱氨酰-tRNA 合成酶,突出了大规模多条件分析在确定细菌变种之间关键表型差异方面的强大功能。