From the ‡Bioanalytics, Institute of Biotechnology, Technische Universität Berlin, 13355 Berlin, Germany.
From the ‡Bioanalytics, Institute of Biotechnology, Technische Universität Berlin, 13355 Berlin, Germany;; §Wellcome Centre for Cell Biology, School of Biological Sciences, University of Edinburgh, Edinburgh EH9 3BF, Scotland, United Kingdom.
Mol Cell Proteomics. 2019 Apr;18(4):786-795. doi: 10.1074/mcp.TIR118.001276. Epub 2019 Jan 16.
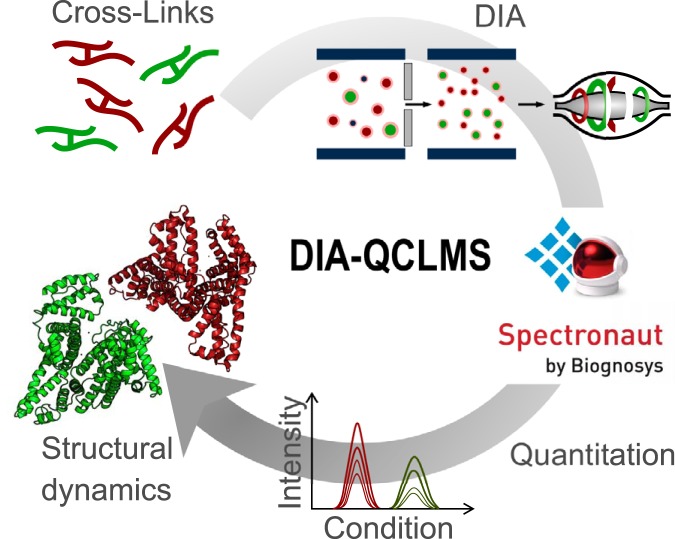
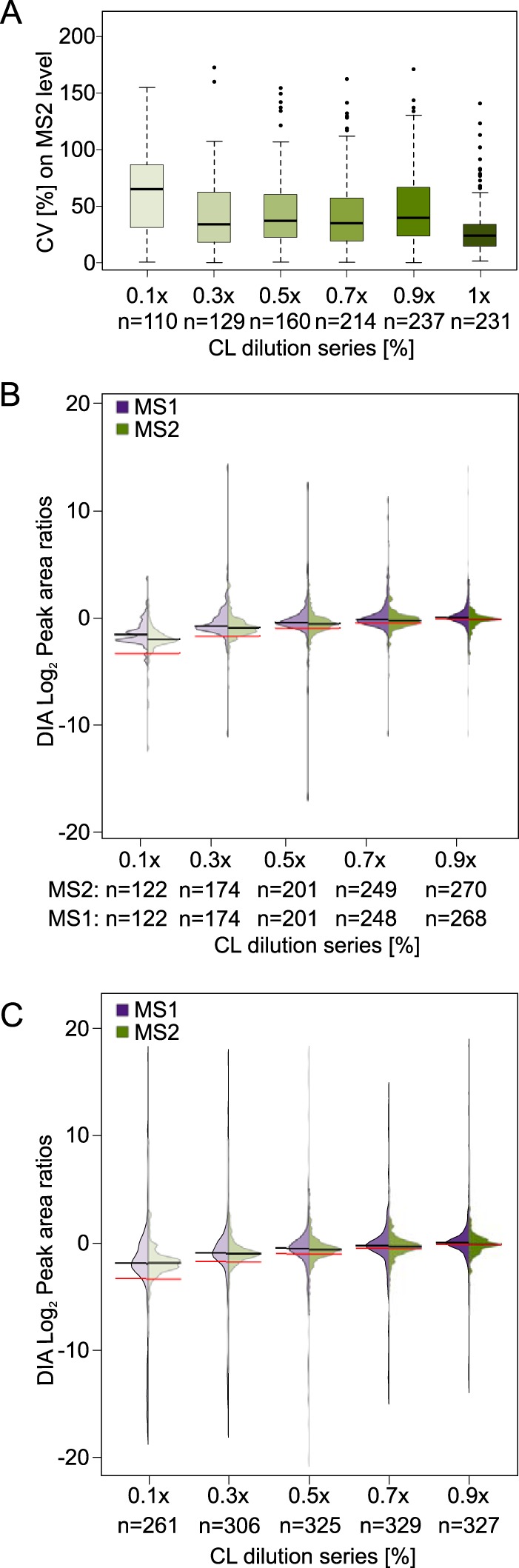
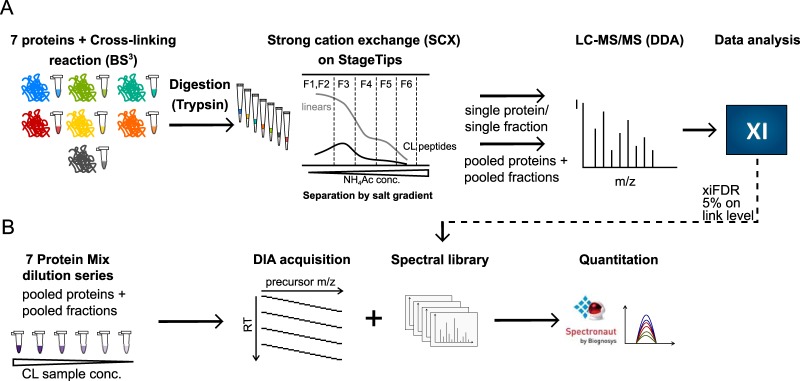
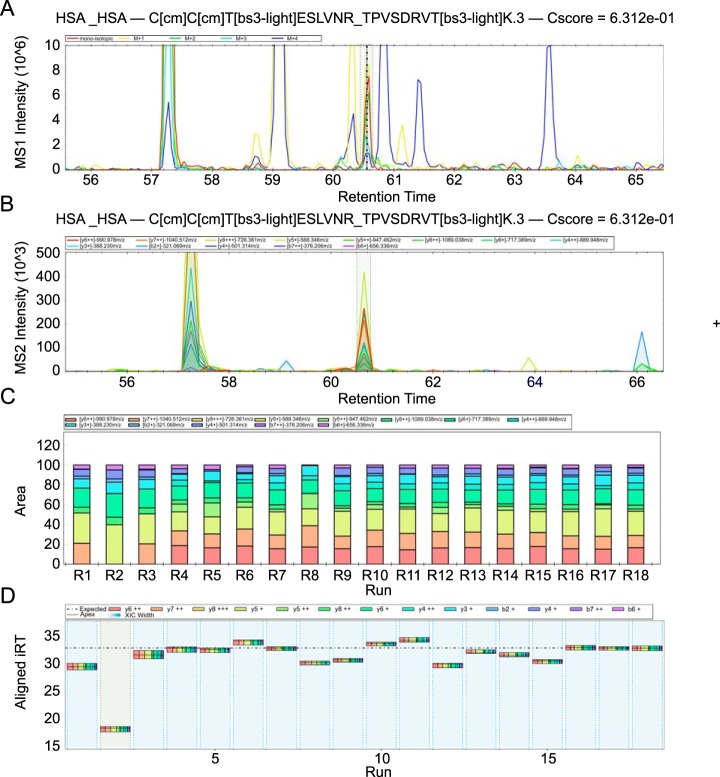
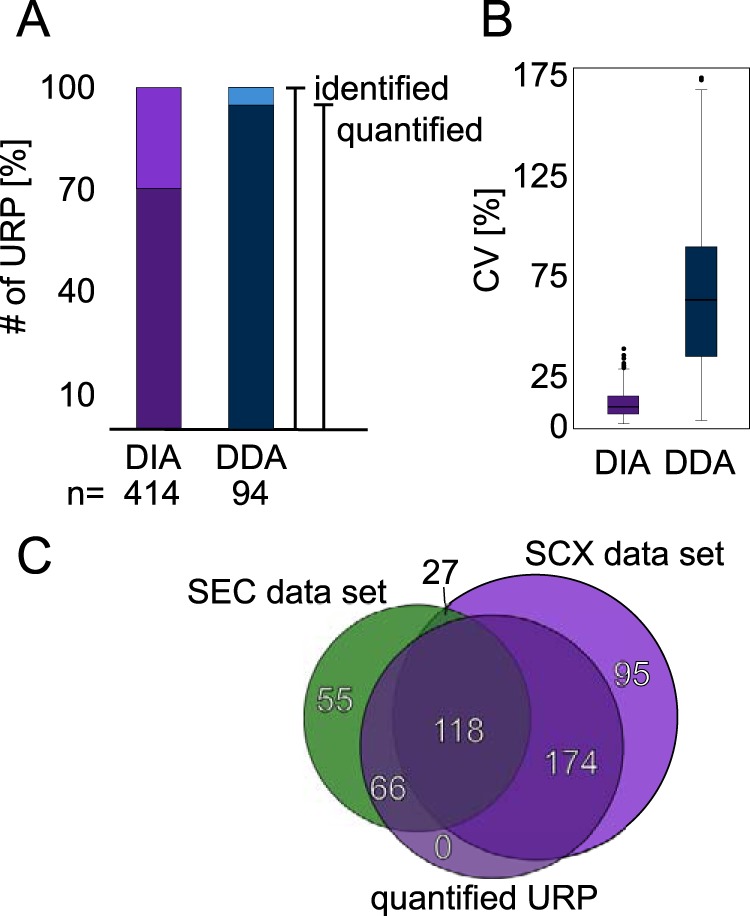
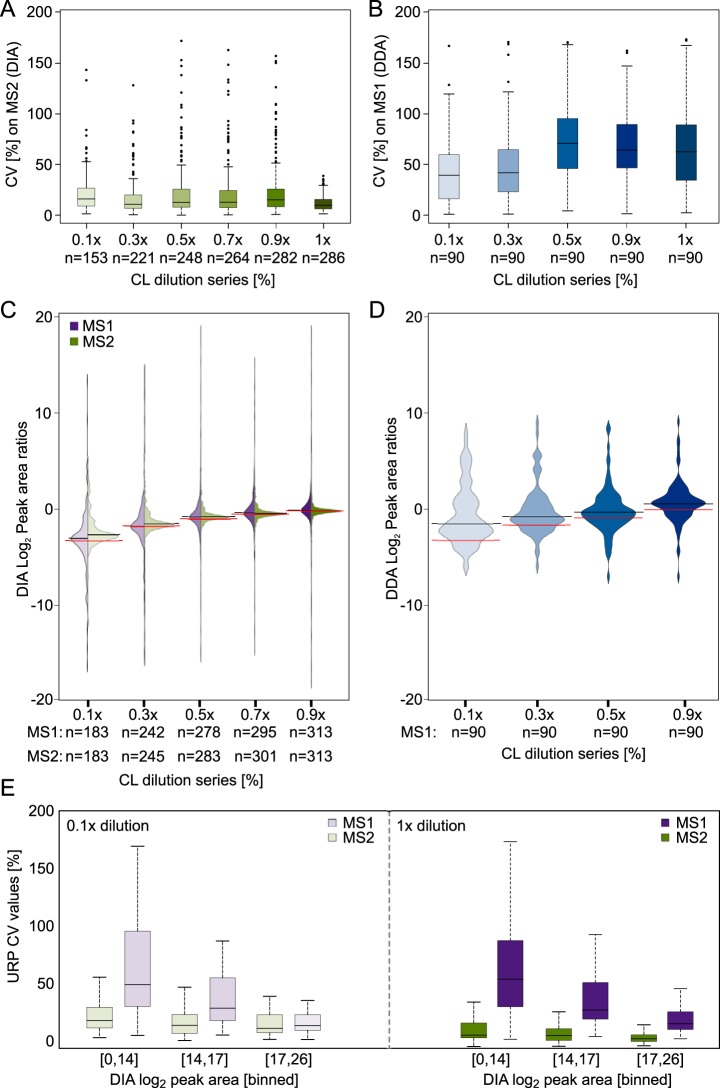
Quantitative cross-linking mass spectrometry (QCLMS) reveals structural detail on altered protein states in solution. On its way to becoming a routine technology, QCLMS could benefit from data-independent acquisition (DIA), which generally enables greater reproducibility than data-dependent acquisition (DDA) and increased throughput over targeted methods. Therefore, here we introduce DIA to QCLMS by extending a widely used DIA software, Spectronaut, to also accommodate cross-link data. A mixture of seven proteins cross-linked with bis[sulfosuccinimidyl] suberate (BS) was used to evaluate this workflow. Out of the 414 identified unique residue pairs, 292 (70%) were quantifiable across triplicates with a coefficient of variation (CV) of 10%, with manual correction of peak selection and boundaries for PSMs in the lower quartile of individual CV values. This compares favorably to DDA where we quantified cross-links across triplicates with a CV of 66%, for a single protein. We found DIA-QCLMS to be capable of detecting changing abundances of cross-linked peptides in complex mixtures, despite the ratio compression encountered when increasing sample complexity through the addition of cell lysate as matrix. In conclusion, the DIA software Spectronaut can now be used in cross-linking and DIA is indeed able to improve QCLMS.
定量交联质谱(QCLMS)揭示了溶液中蛋白质状态改变的结构细节。在成为常规技术的过程中,QCLMS 可以受益于数据独立采集(DIA),DIA 通常比数据依赖采集(DDA)具有更高的重现性,并且比靶向方法具有更高的通量。因此,我们通过扩展广泛使用的 DIA 软件 Spectronaut 来将 DIA 引入 QCLMS,使其也能够适应交联数据。使用混合了七种用双[磺基琥珀酰亚胺]酯(BS)交联的蛋白质来评估此工作流程。在鉴定的 414 个独特残基对中,有 292 个(70%)可在三重复中定量,变异系数(CV)为 10%,通过手动校正峰选择和 PSM 边界来纠正个别 CV 值处于较低四分位数的情况。与 DDA 相比,我们在单个蛋白质中定量了三重复的交联,CV 为 66%,这是有利的。我们发现,尽管通过添加细胞裂解物作为基质来增加样品复杂性会导致比率压缩,但 DIA-QCLMS 仍能够检测复杂混合物中交联肽丰度的变化。总之,交联和 DIA 现在可以使用 DIA 软件 Spectronaut,并且 DIA 确实能够改进 QCLMS。