Clark Lindsay V, Lipka Alexander E, Sacks Erik J
Department of Crop Sciences, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801
Department of Crop Sciences, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801.
G3 (Bethesda). 2019 Mar 7;9(3):663-673. doi: 10.1534/g3.118.200913.
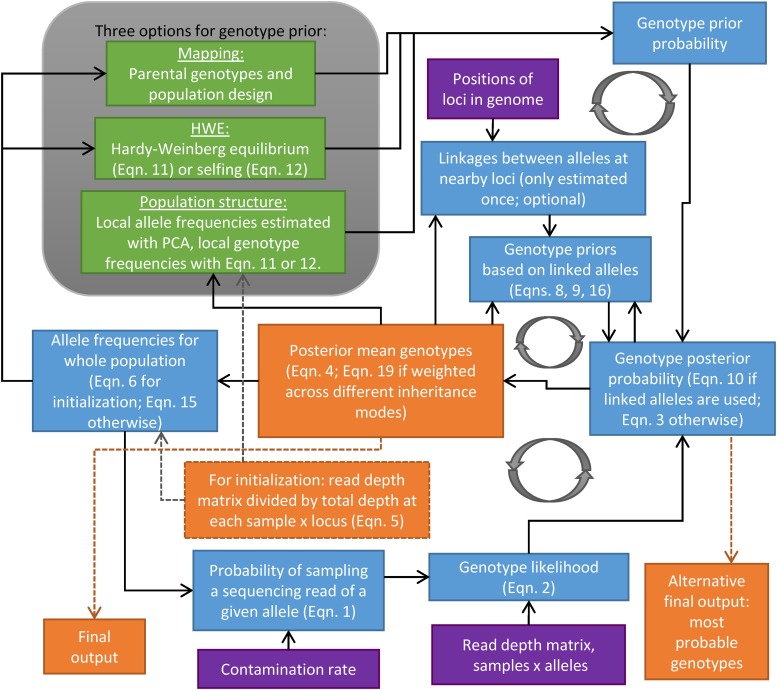
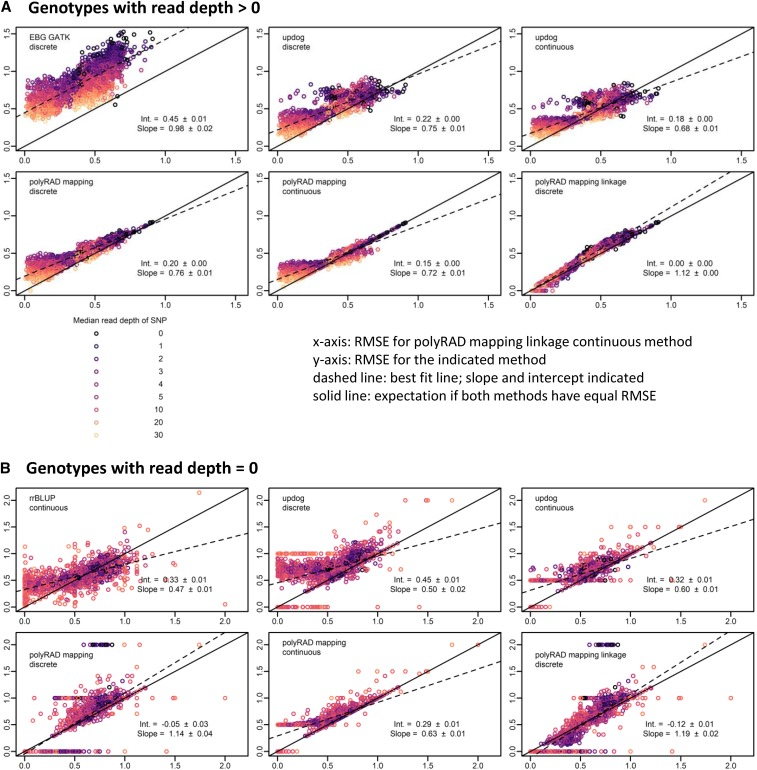
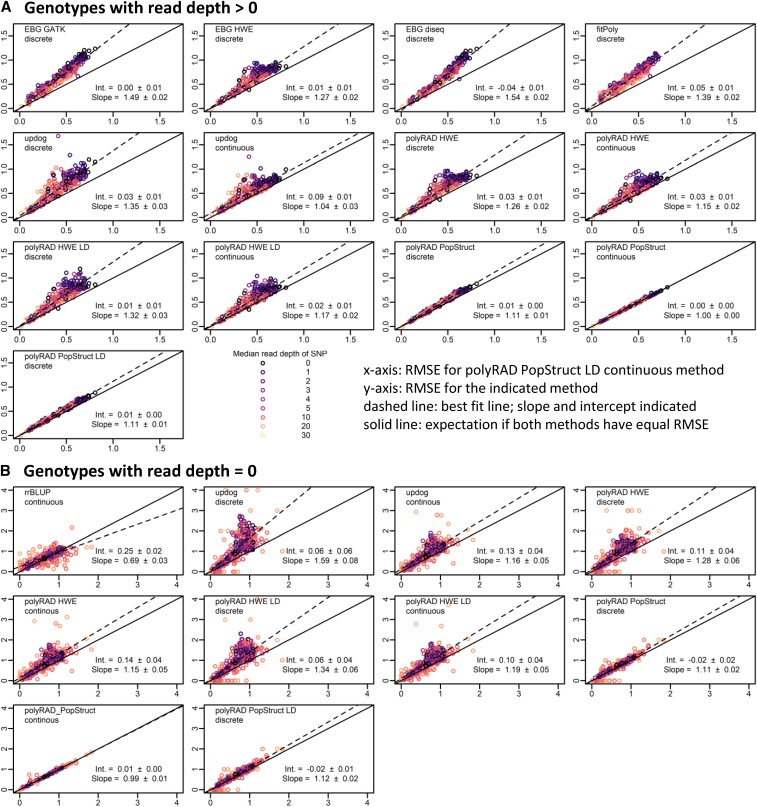
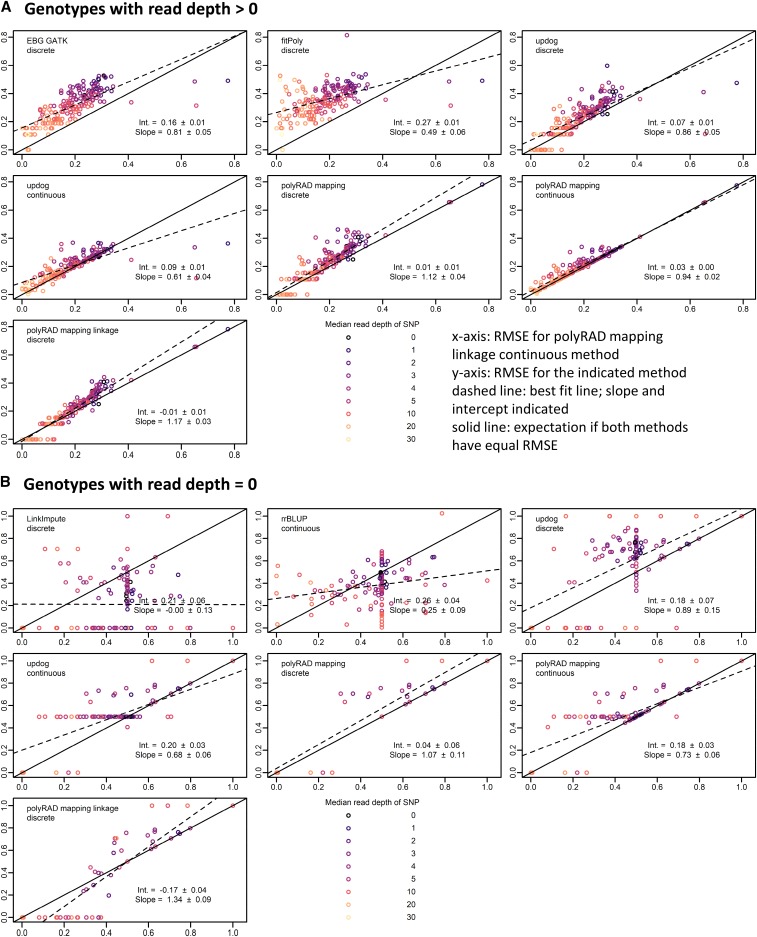
Low or uneven read depth is a common limitation of genotyping-by-sequencing (GBS) and restriction site-associated DNA sequencing (RAD-seq), resulting in high missing data rates, heterozygotes miscalled as homozygotes, and uncertainty of allele copy number in heterozygous polyploids. Bayesian genotype calling can mitigate these issues, but previously has only been implemented in software that requires a reference genome or uses priors that may be inappropriate for the population. Here we present several novel Bayesian algorithms that estimate genotype posterior probabilities, all of which are implemented in a new R package, polyRAD. Appropriate priors can be specified for mapping populations, populations in Hardy-Weinberg equilibrium, or structured populations, and in each case can be informed by genotypes at linked markers. The polyRAD software imports read depth from several existing pipelines, and outputs continuous or discrete numerical genotypes suitable for analyses such as genome-wide association and genomic prediction.
低读深度或不均匀读深度是简化基因组测序(GBS)和限制性位点相关DNA测序(RAD-seq)的常见局限,会导致高缺失数据率、杂合子被误称为纯合子,以及杂合多倍体中等位基因拷贝数的不确定性。贝叶斯基因型分型可以缓解这些问题,但以前仅在需要参考基因组或使用可能不适用于该群体的先验信息的软件中实现。在此,我们提出了几种估计基因型后验概率的新型贝叶斯算法,所有这些算法都在一个新的R包polyRAD中实现。可以为作图群体、处于哈迪-温伯格平衡的群体或结构化群体指定合适的先验信息,并且在每种情况下都可以通过连锁标记处的基因型来提供信息。polyRAD软件从几个现有流程中导入读深度,并输出适用于全基因组关联分析和基因组预测等分析的连续或离散数字基因型。