Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland.
Department of Chemistry and Biochemistry, Utah State University, Logan, UT 84322-0300, USA.
Molecules. 2019 Jan 21;24(2):376. doi: 10.3390/molecules24020376.
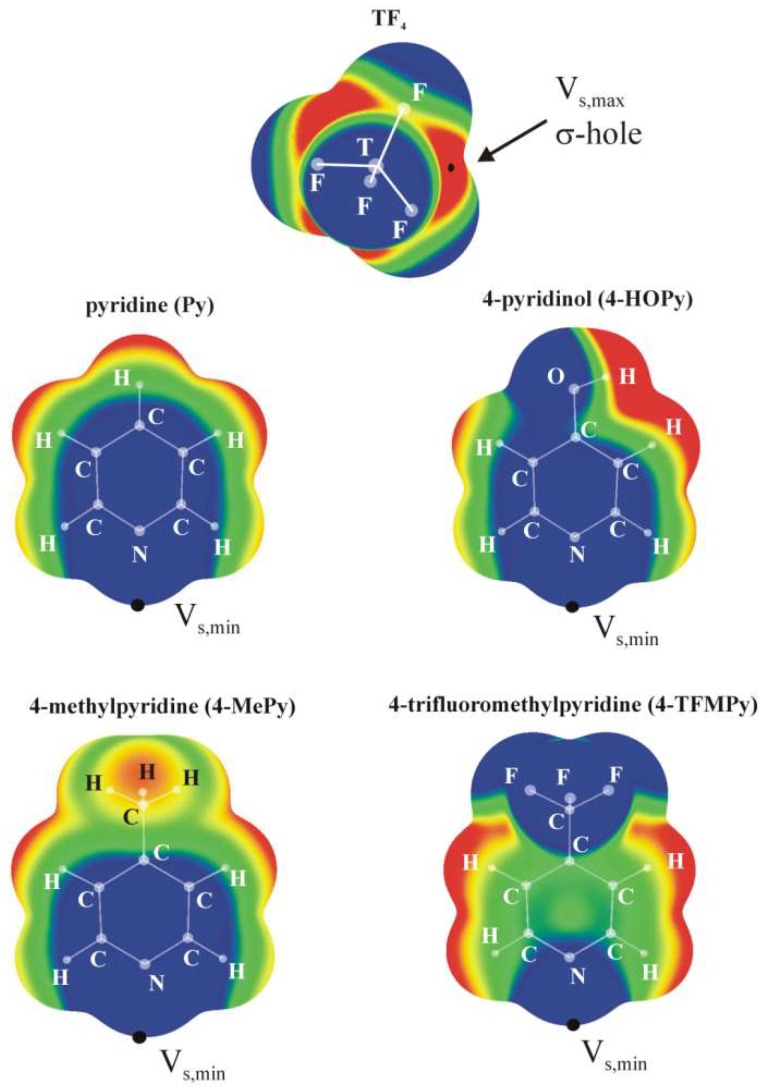
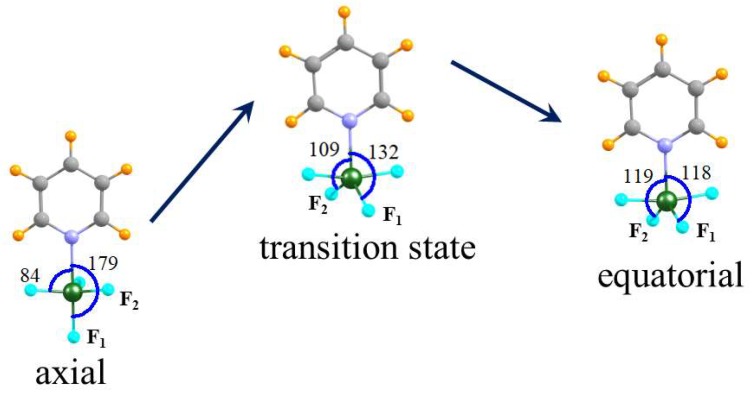
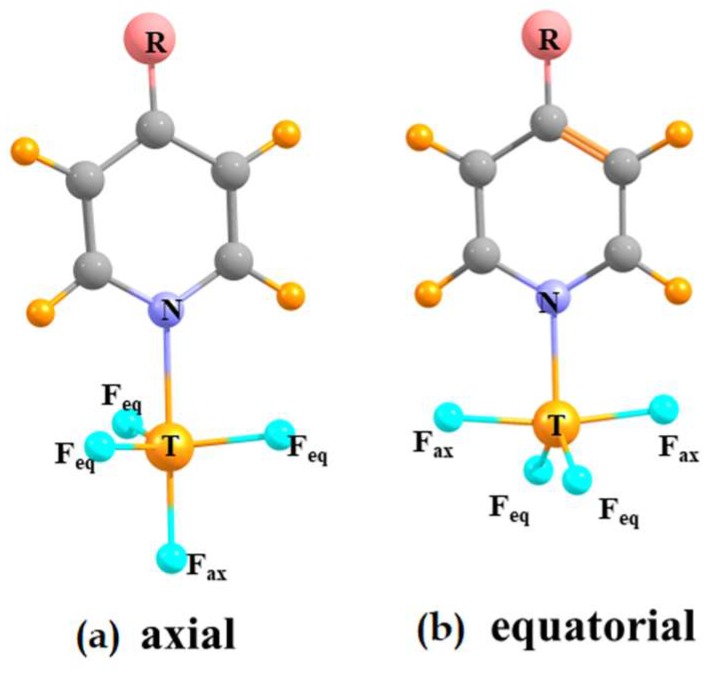
When an N-base approaches the tetrel atom of TF₄ (T = Si, Ge, Sn) the latter molecule deforms from a tetrahedral structure in the monomer to a trigonal bipyramid. The base can situate itself at either an axial or equatorial position, leading to two different equilibrium geometries. The interaction energies are considerably larger for the equatorial structures, up around 50 kcal/mol, which also have a shorter R(T··N) separation. On the other hand, the energy needed to deform the tetrahedral monomer into the equatorial structure is much higher than the equivalent deformation energy in the axial dimer. When these two opposite trends are combined, it is the axial geometry which is somewhat more stable than the equatorial, yielding binding energies in the 8⁻34 kcal/mol range. There is a clear trend of increasing interaction energy as the tetrel atom grows larger: Si < Ge < Sn, a pattern which is accentuated for the binding energies.
当 N 基底接近 TF₄(T = Si、Ge、Sn)中的四配位原子时,后者的分子会从单体的四面体结构变形为三角双锥。基底可以位于轴向或赤道位置,导致两种不同的平衡几何结构。对于赤道结构,相互作用能要大得多,约为 50 kcal/mol,而且 R(T··N) 间隔也更短。另一方面,将四面体单体变形为赤道结构所需的能量远高于轴向二聚体的等效变形能。当这两种相反的趋势结合在一起时,轴向几何结构比赤道结构稍微稳定一些,结合能在 8⁻34 kcal/mol 的范围内。随着四配位原子的增大,相互作用能明显增大:Si < Ge < Sn,对于结合能来说,这种模式更为明显。