Cardiovascular Research Centre, Cardiovascular Genetics and Genomics group at Royal Brompton Hospital and Harefield NHS Foundation Trust, Sydney Street, London, SW3 6NP, UK.
National Heart and Lung Institute, Imperial College London, London, UK.
Genome Med. 2019 Jan 29;11(1):5. doi: 10.1186/s13073-019-0616-z.
International guidelines for variant interpretation in Mendelian disease set stringent criteria to report a variant as (likely) pathogenic, prioritising control of false-positive rate over test sensitivity and diagnostic yield. Genetic testing is also more likely informative in individuals with well-characterised variants from extensively studied European-ancestry populations. Inherited cardiomyopathies are relatively common Mendelian diseases that allow empirical calibration and assessment of this framework.
We compared rare variants in large hypertrophic cardiomyopathy (HCM) cohorts (up to 6179 cases) to reference populations to identify variant classes with high prior likelihoods of pathogenicity, as defined by etiological fraction (EF). We analysed the distribution of variants using a bespoke unsupervised clustering algorithm to identify gene regions in which variants are significantly clustered in cases.
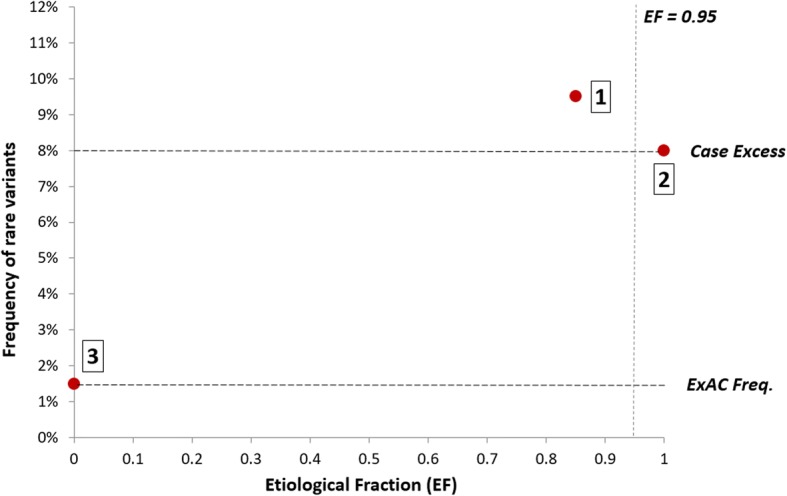
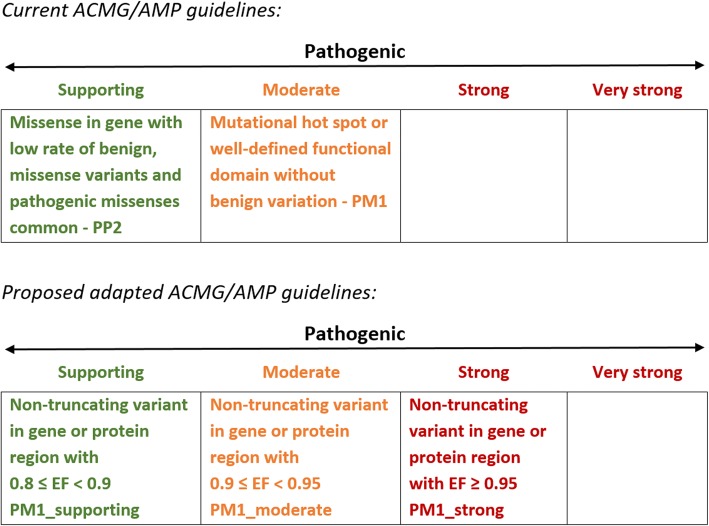
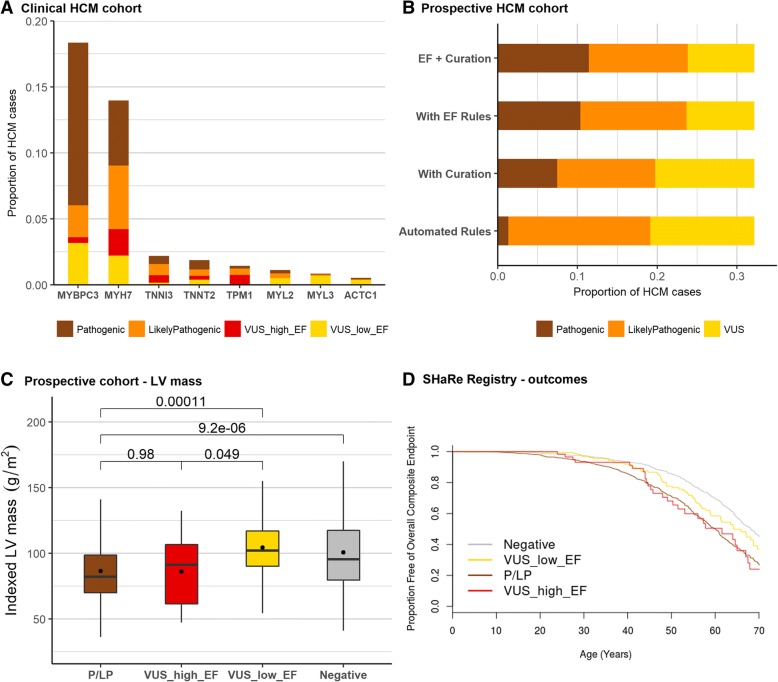
Analysis of variant distribution identified regions in which variants are significantly enriched in cases and variant location was a better discriminator of pathogenicity than generic computational functional prediction algorithms. Non-truncating variant classes with an EF ≥ 0.95 were identified in five established HCM genes. Applying this approach leads to an estimated 14-20% increase in cases with actionable HCM variants, i.e. variants classified as pathogenic/likely pathogenic that might be used for predictive testing in probands' relatives.
When found in a patient confirmed to have disease, novel variants in some genes and regions are empirically shown to have a sufficiently high probability of pathogenicity to support a "likely pathogenic" classification, even without additional segregation or functional data. This could increase the yield of high confidence actionable variants, consistent with the framework and recommendations of current guidelines. The techniques outlined offer a consistent and unbiased approach to variant interpretation for Mendelian disease genetic testing. We propose adaptations to ACMG/AMP guidelines to incorporate such evidence in a quantitative and transparent manner.
孟德尔疾病变异解释的国际指南设定了严格的标准,将变异报告为(可能)致病性,优先控制假阳性率,而不是提高检测敏感性和诊断率。遗传检测在来自广泛研究的欧洲血统人群中具有特征明确的变异个体中更有可能具有信息性。遗传性心肌病是相对常见的孟德尔疾病,可以对该框架进行经验校准和评估。
我们将大型肥厚型心肌病(HCM)队列(多达 6179 例)中的罕见变异与参考人群进行比较,以确定具有高致病性先验可能性的变异类别,如病因分数(EF)所定义。我们使用定制的无监督聚类算法分析变异的分布,以识别在病例中变异明显聚类的基因区域。
分析变异分布确定了在病例中变异明显富集的区域,并且变异位置比通用计算功能预测算法更好地区分致病性。在五个已建立的 HCM 基因中确定了 EF≥0.95 的非截断变异类别。应用此方法可导致具有可操作 HCM 变异的病例估计增加 14-20%,即被归类为致病性/可能致病性的变异,这些变异可能用于在先证者亲属中进行预测性检测。
当在已确诊患有疾病的患者中发现新变异时,一些基因和区域的变异被经验证明具有足够高的致病性概率,足以支持“可能致病性”的分类,即使没有额外的分离或功能数据。这可以增加高置信度可操作变异的产量,与当前指南的框架和建议一致。概述的技术为孟德尔疾病基因检测的变异解释提供了一种一致且无偏的方法。我们建议对 ACMG/AMP 指南进行修改,以定量和透明的方式纳入此类证据。