Sheffield Institute for Translational Neuroscience (SITraN), University of Sheffield, 385 Glossop Road, Sheffield, UK.
The Living Systems Institute, University of Exeter, Stocker Road, Exeter, UK.
Brain. 2019 Mar 1;142(3):586-605. doi: 10.1093/brain/awy353.
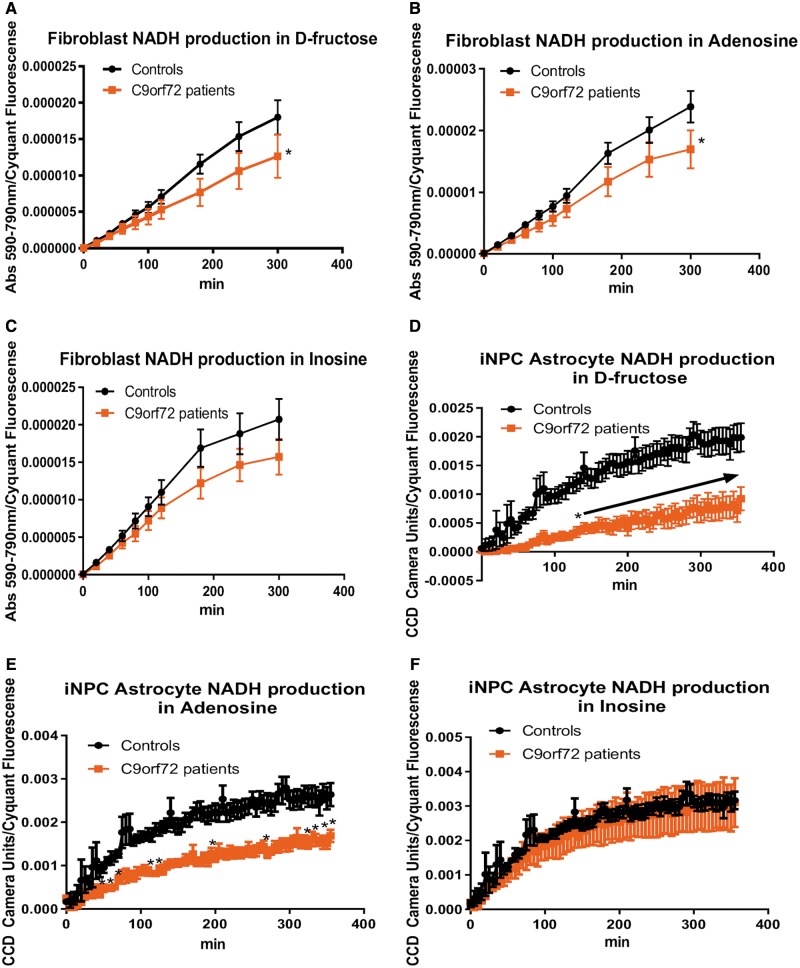
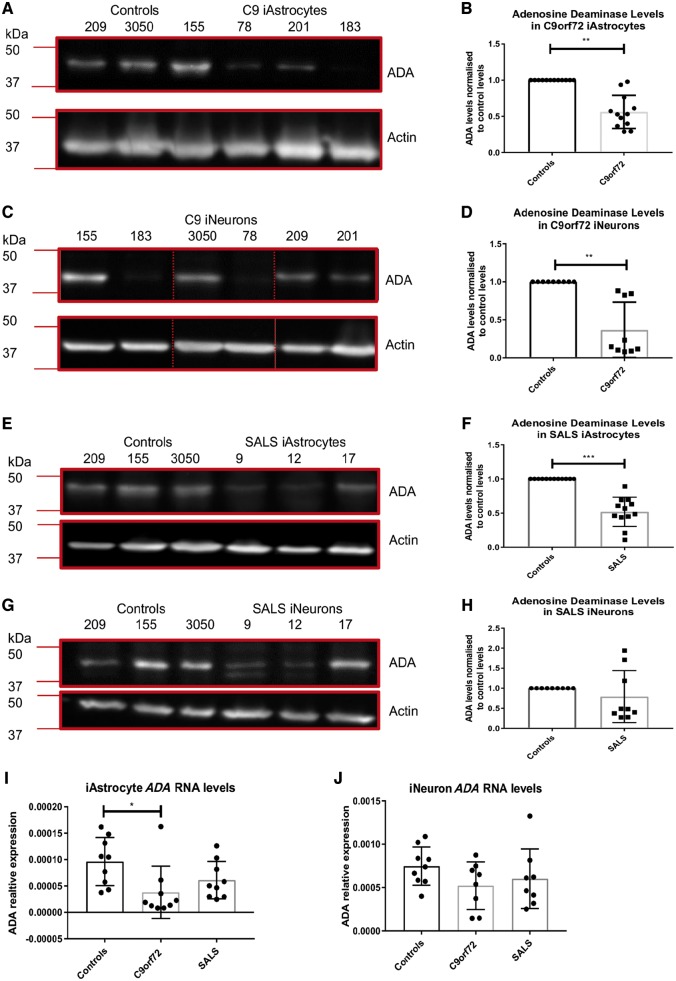
As clinical evidence supports a negative impact of dysfunctional energy metabolism on the disease progression in amyotrophic lateral sclerosis, it is vital to understand how the energy metabolic pathways are altered and whether they can be restored to slow disease progression. Possible approaches include increasing or rerouting catabolism of alternative fuel sources to supplement the glycolytic and mitochondrial pathways such as glycogen, ketone bodies and nucleosides. To analyse the basis of the catabolic defect in amyotrophic lateral sclerosis we used a novel phenotypic metabolic array. We profiled fibroblasts and induced neuronal progenitor-derived human induced astrocytes from C9orf72 amyotrophic lateral sclerosis patients compared to normal controls, measuring the rates of production of reduced nicotinamide adenine dinucleotides from 91 potential energy substrates. This approach shows for the first time that C9orf72 human induced astrocytes and fibroblasts have an adenosine to inosine deamination defect caused by reduction of adenosine deaminase, which is also observed in induced astrocytes from sporadic patients. Patient-derived induced astrocyte lines were more susceptible to adenosine-induced toxicity, which could be mimicked by inhibiting adenosine deaminase in control lines. Furthermore, adenosine deaminase inhibition in control induced astrocytes led to increased motor neuron toxicity in co-cultures, similar to the levels observed with patient derived induced astrocytes. Bypassing metabolically the adenosine deaminase defect by inosine supplementation was beneficial bioenergetically in vitro, increasing glycolytic energy output and leading to an increase in motor neuron survival in co-cultures with induced astrocytes. Inosine supplementation, in combination with modulation of the level of adenosine deaminase may represent a beneficial therapeutic approach to evaluate in patients with amyotrophic lateral sclerosis.
由于临床证据支持能量代谢功能障碍对肌萎缩侧索硬化症疾病进展的负面影响,因此了解能量代谢途径如何改变以及是否可以恢复以减缓疾病进展至关重要。可能的方法包括增加或重新引导替代燃料的分解代谢以补充糖酵解和线粒体途径,如糖原、酮体和核苷。为了分析肌萎缩侧索硬化症中分解代谢缺陷的基础,我们使用了一种新型表型代谢组学。我们对 C9orf72 肌萎缩侧索硬化症患者的成纤维细胞和诱导神经元祖细胞衍生的人类诱导星形胶质细胞进行了分析,并与正常对照进行了比较,测量了 91 种潜在能量底物产生还原型烟酰胺腺嘌呤二核苷酸的速率。这种方法首次表明,C9orf72 人类诱导星形胶质细胞和成纤维细胞存在由于腺苷脱氨酶减少引起的腺苷到肌苷脱氨缺陷,这在散发性患者的诱导星形胶质细胞中也观察到。患者来源的诱导星形胶质细胞系对腺苷诱导的毒性更敏感,在对照系中抑制腺苷脱氨酶可模拟这种毒性。此外,在对照诱导星形胶质细胞中抑制腺苷脱氨酶会导致共培养物中运动神经元毒性增加,类似于与患者来源的诱导星形胶质细胞观察到的水平。通过肌苷补充绕过腺苷脱氨酶缺陷的代谢途径在体外具有有益的生物能量学作用,增加糖酵解能量输出,并导致与诱导星形胶质细胞共培养物中的运动神经元存活增加。肌苷补充与腺苷脱氨酶水平的调节相结合可能代表一种有益的治疗方法,可在肌萎缩侧索硬化症患者中进行评估。