Nicolescu Ramona C, Al-Khawaga Sara, Minassian Berge A, Hussain Khalid
Division of Endocrinology and Diabetes, Department of Pediatrics, University of Liège, Centre Hospitalier Régional de la Citadelle, Liège, Belgium.
Division of Endocrinology, Department of Pediatrics, Sidra Medicine Outpatient Clinic, Doha, Qatar.
Front Pediatr. 2019 Jan 16;6:424. doi: 10.3389/fped.2018.00424. eCollection 2018.
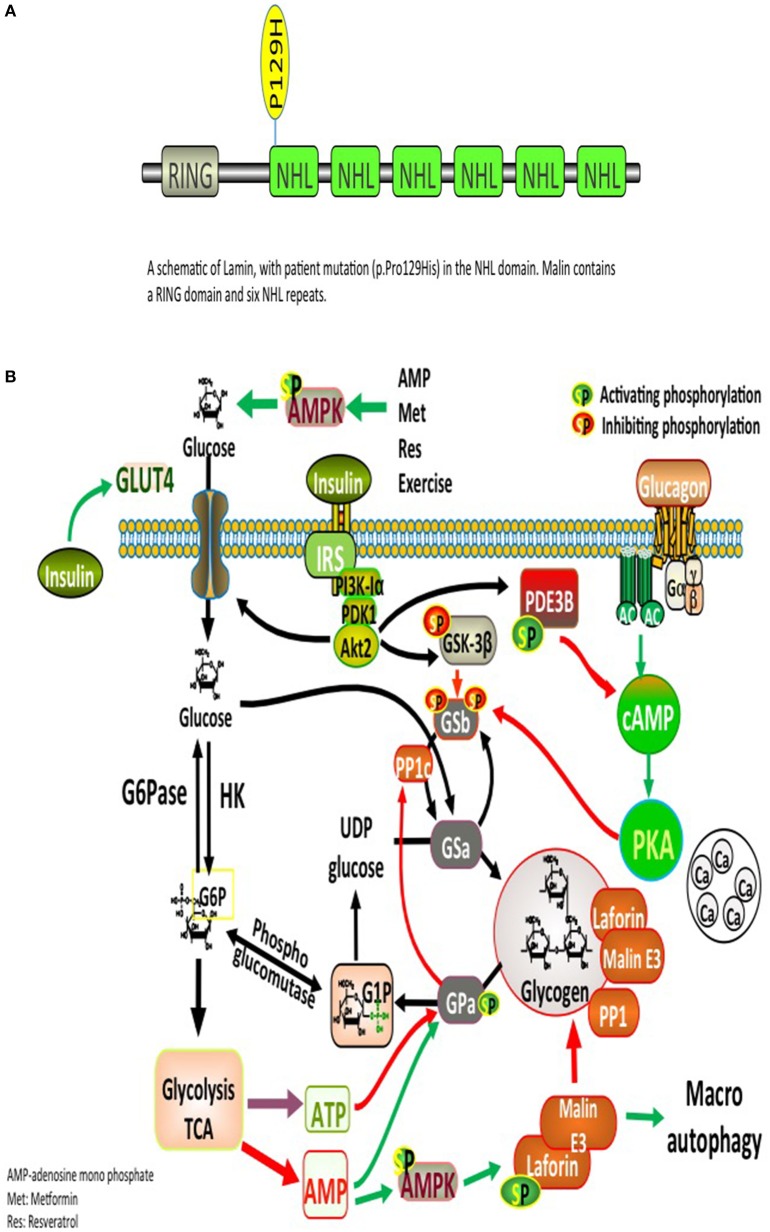
Lafora disease (LD) is a rare autosomal recessive disorder characterized by progressive myoclonic epilepsy followed by continuous neurological decline, culminating in death within 10 years. LD leads to accumulation of insoluble, abnormal, glycogen-like structures called Lafora bodies (LBs). It is caused by mutations in the gene encoding glycogen phosphatase ( or the E3 ubiquitin ligase malin (. These two proteins are involved in an intricate, however, incompletely elucidated pathway governing glycogen metabolism. The formation of EPM2A and malin signaling complex promotes the ubiquitination of proteins participating in glycogen metabolism, where dysfunctional mutations lead to the formation of LBs. Herein, we describe a 13-years-old child with LD due to a (c.386C > A, p.Pro129His) mutation, who has developed diabetes mellitus and was treated with metformin. We discuss how basic mechanisms of LD could be linked to β-cell dysfunction and insulin resistance.
拉福拉病(LD)是一种罕见的常染色体隐性疾病,其特征为进行性肌阵挛性癫痫,随后出现持续的神经功能衰退,最终在10年内死亡。LD会导致称为拉福拉小体(LBs)的不溶性、异常、糖原样结构的积累。它是由编码糖原磷酸酶(或E3泛素连接酶malin)的基因突变引起的。这两种蛋白质参与了一个复杂但尚未完全阐明的糖原代谢调控途径。EPM2A和malin信号复合物的形成促进了参与糖原代谢的蛋白质的泛素化,功能失调的突变会导致LBs的形成。在此,我们描述了一名因(c.386C>A,p.Pro129His)突变而患有LD的13岁儿童,该儿童已患糖尿病并接受二甲双胍治疗。我们讨论了LD的基本机制如何与β细胞功能障碍和胰岛素抵抗相关联。