Cole Joby, Hanson Eleanor J, James David C, Dockrell David H, Dickman Mark J
Department of Infection, Immunity and Cardiovascular Diseases, University of Sheffield, UK.
Department of Chemical and Biological Engineering, University of Sheffield, UK.
Rapid Commun Mass Spectrom. 2019 May 30;33(10):897-906. doi: 10.1002/rcm.8401.
Histone post-translational modifications (PTMs) play key roles in regulating eukaryotic gene expression. Mass spectrometry (MS) has emerged as a powerful method to characterize and quantify histone PTMs as it allows unbiased identification and quantification of multiple histone PTMs including combinations of the modifications present.
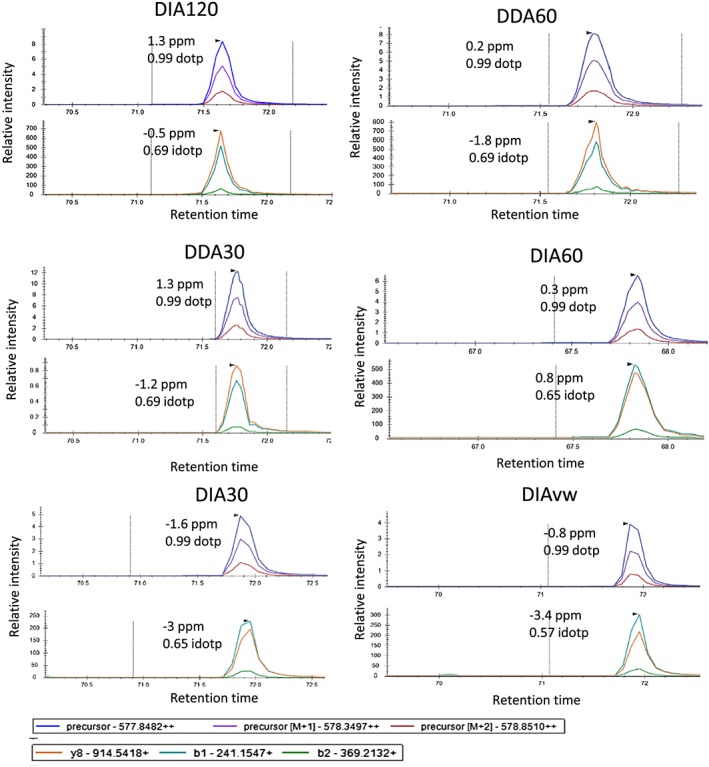
In this study we compared a range of data-acquisition methods for the identification and quantification of the histone PTMs using a Q Exactive HF Orbitrap. We compared three different data-dependent analysis (DDA) methods with MS2 resolutions of 120K, 60K, 30K. We also compared a range of data-independent analysis (DIA) methods using MS2 isolation windows of 20 m/z and DIAvw to identify and quantify histone PTMs in Chinese hamster ovary (CHO) cells.
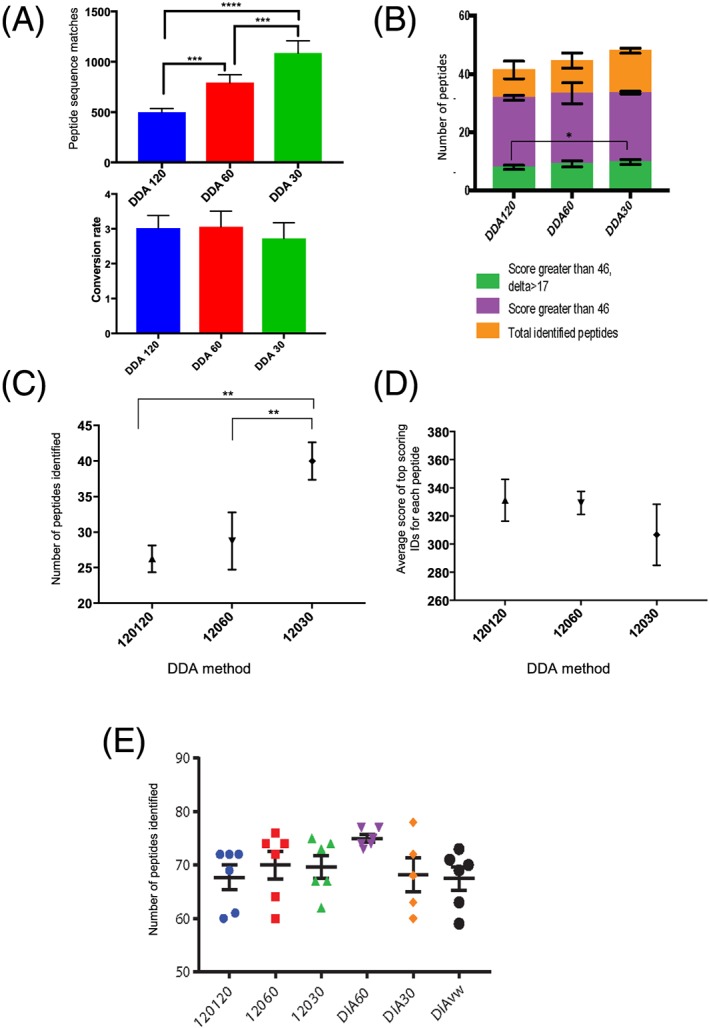
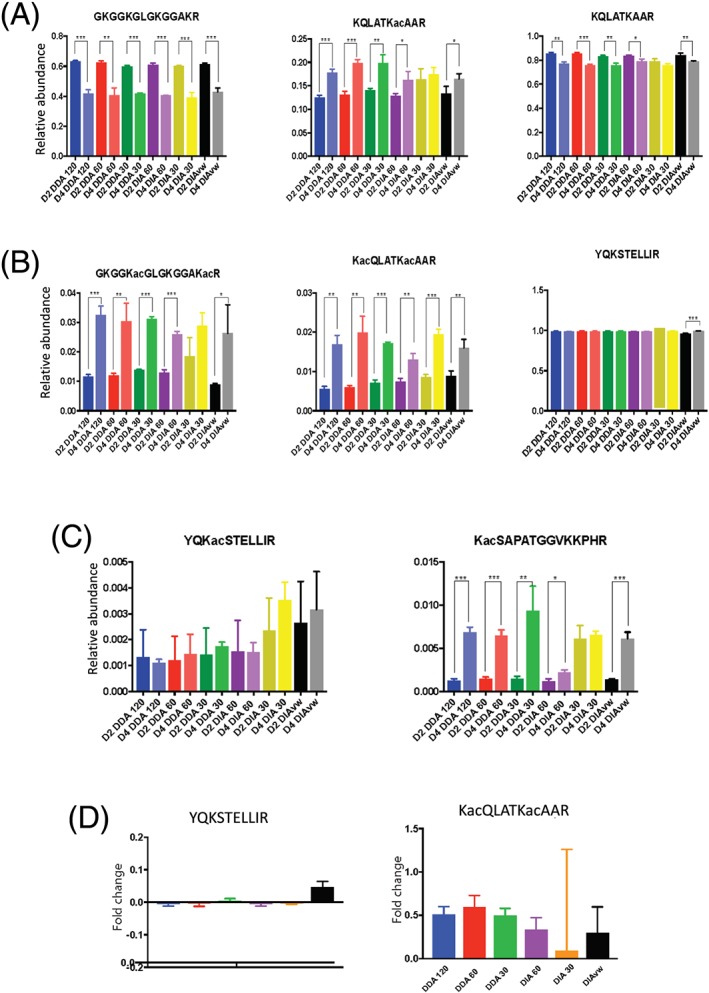
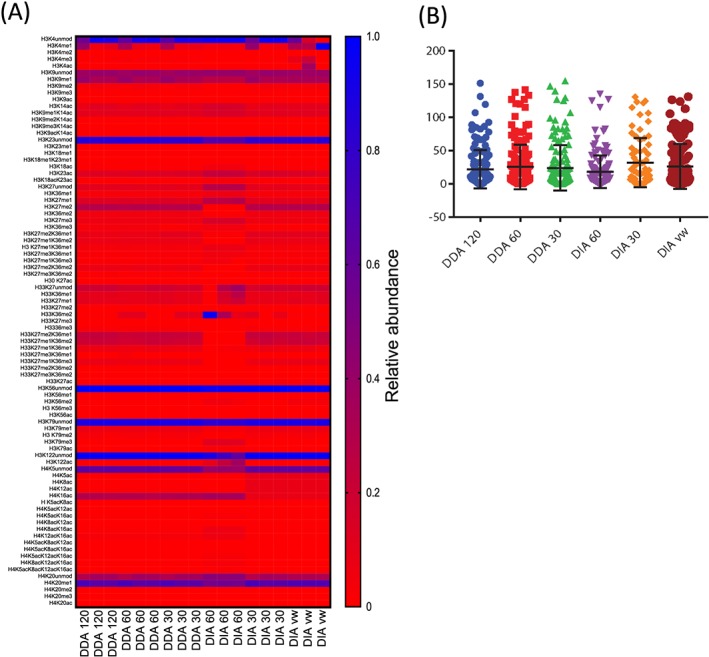
The increased number of MS2 scans afforded by the lower resolution methods resulted in a higher number of queries, peptide sequence matches (PSMs) and a higher number of peptide proteoforms identified with a Mascot Ion score greater than 46. No difference in the proportion of peptide proteoforms with Delta scores >17 was observed. Lower coefficients of variation (CVs) were obtained in the DIA MS1 60 K MS2 30 K 20 m/z isolation windows compared with the other data-acquisition methods.
We observed that DIA which offers advantages in flexibility and identification of isobaric peptide proteoforms performs as well as DDA in the analysis of histone PTMs. We were able to identify 71 modified histone peptides for histone H3 and H4 and quantified 64 across each of the different acquisition methods.
组蛋白翻译后修饰(PTMs)在调节真核基因表达中起关键作用。质谱(MS)已成为表征和定量组蛋白PTMs的有力方法,因为它能够对多种组蛋白PTMs进行无偏倚的鉴定和定量,包括存在的修饰组合。
在本研究中,我们使用Q Exactive HF Orbitrap比较了一系列用于鉴定和定量组蛋白PTMs的数据采集方法。我们比较了三种不同的数据依赖分析(DDA)方法,其二级质谱分辨率分别为120K、60K、30K。我们还比较了一系列数据独立分析(DIA)方法,使用20 m/z的二级质谱隔离窗口和DIAvw来鉴定和定量中国仓鼠卵巢(CHO)细胞中的组蛋白PTMs。
较低分辨率方法提供的二级质谱扫描次数增加,导致查询次数、肽序列匹配(PSM)次数增加,以及通过Mascot离子得分大于46鉴定的肽蛋白异构体数量增加。未观察到Delta得分>17的肽蛋白异构体比例存在差异。与其他数据采集方法相比,在DIA一级质谱60K、二级质谱30K、20 m/z隔离窗口中获得了更低的变异系数(CV)。
我们观察到,在灵活性和等压肽蛋白异构体鉴定方面具有优势的DIA在组蛋白PTMs分析中与DDA表现相当。我们能够鉴定出71种组蛋白H3和H4的修饰组蛋白肽,并通过每种不同的采集方法对64种进行了定量。