Department of Genetics and Genomic Sciences and Medicine, Icahn School of Medicine at Mount Sinai, New York, NY 10029, United States.
Mol Genet Metab. 2019 Nov;128(3):298-303. doi: 10.1016/j.ymgme.2019.01.020. Epub 2019 Jan 24.
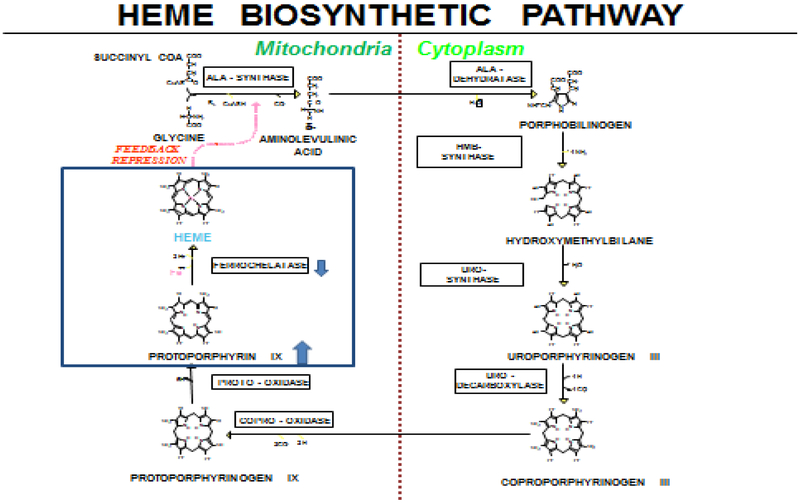
Erythropoietic Protoporphyria (EPP) and X-linked Protoporphyria (XLP) are rare, genetic photodermatoses resulting from defects in enzymes of the heme-biosynthetic pathway. EPP results from the partial deficiency of ferrochelatase, and XLP results from gain-of-function mutations in erythroid specific ALAS2. Both disorders result in the accumulation of erythrocyte protoporphyrin, which is released in the plasma and taken up by the liver and vascular endothelium. The accumulated protoporphyrin is activated by sunlight exposure, generating singlet oxygen radical reactions leading to tissue damage and excruciating pain. About 2-5% of patients develop clinically significant liver dysfunction due to protoporphyrin deposition in bile and/or hepatocytes which can advance to cholestatic liver failure requiring transplantation. Clinically these patients present with acute, severe, non-blistering phototoxicity within minutes of sun-exposure. Anemia is seen in about 47% of patients and about 27% of patients will develop abnormal serum aminotransferases. The diagnosis of EPP and XLP is made by detection of markedly increased erythrocyte protoporphyrin levels with a predominance of metal-free protoporphyrin. Genetic testing by sequencing the FECH or ALAS2 gene confirms the diagnosis. Treatment is limited to sun-protection and there are no currently available FDA-approved therapies for these disorders. Afamelanotide, a synthetic analogue of α-melanocyte stimulating hormone was found to increase pain-free sun exposure and improve quality of life in adults with EPP. It has been approved for use in the European Union since 2014 and is not available in the U.S. In addition to the development of effective therapeutics, future studies are needed to establish the role of iron and the risks related to the development of hepatopathy in these patients.
红细胞生成性原卟啉症(EPP)和 X 连锁原卟啉症(XLP)是罕见的遗传性光皮病,由血红素生物合成途径中酶的缺陷引起。EPP 是由于亚铁螯合酶的部分缺乏引起的,而 XLP 是由于红系特异性 ALAS2 的功能获得性突变引起的。两种疾病都会导致红细胞原卟啉的积累,原卟啉会释放到血浆中,并被肝脏和血管内皮细胞摄取。积累的原卟啉在阳光照射下被激活,产生单线态氧自由基反应,导致组织损伤和剧痛。大约 2-5%的患者会因胆汁和/或肝细胞中原卟啉的沉积而出现明显的肝功能障碍,进而进展为需要移植的胆汁淤积性肝衰竭。临床上,这些患者在阳光照射后几分钟内会出现急性、严重、非水疱性光毒性。约 47%的患者会出现贫血,约 27%的患者会出现血清转氨酶异常。EPP 和 XLP 的诊断是通过检测红细胞中原卟啉水平明显升高,以无金属原卟啉为主。通过测序 FECH 或 ALAS2 基因进行基因检测可确诊。治疗仅限于防晒,目前尚无针对这些疾病的 FDA 批准的治疗方法。阿法美拉诺肽是一种α-促黑素细胞激素的合成类似物,被发现可以增加 EPP 成人无疼痛的阳光暴露时间,并改善生活质量。自 2014 年以来,它已在欧盟获得批准使用,在美国不可用。除了开发有效的治疗方法外,还需要进行未来的研究,以确定这些患者中铁的作用和与肝病变发展相关的风险。