Li Haixia, Obligacion Jennifer V, Chirik Paul J, Hall Michael B
Department of Chemistry, Texas A&M University, College Station, Texas 77843, USA.
Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA.
ACS Catal. 2018 Nov 2;8(11):10606-10618. doi: 10.1021/acscatal.8b03146. Epub 2018 Oct 17.
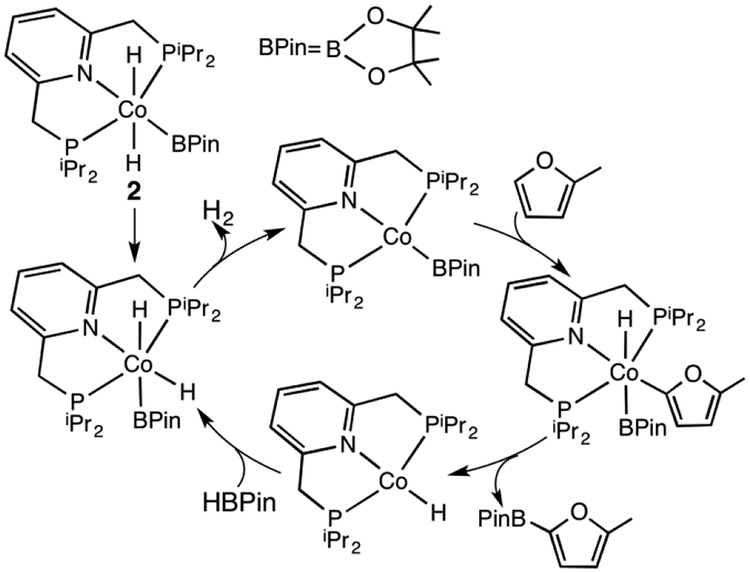
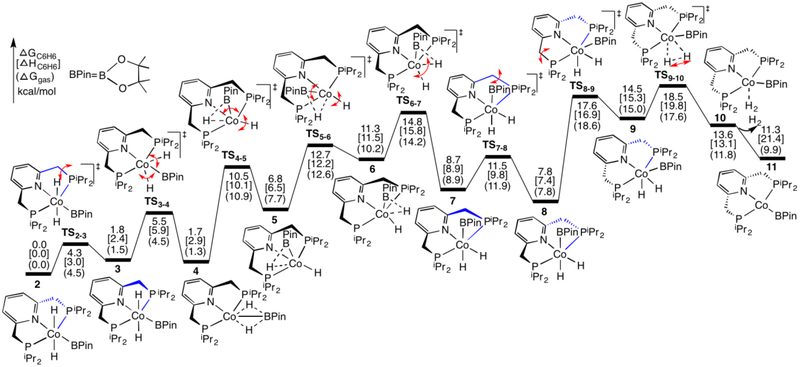
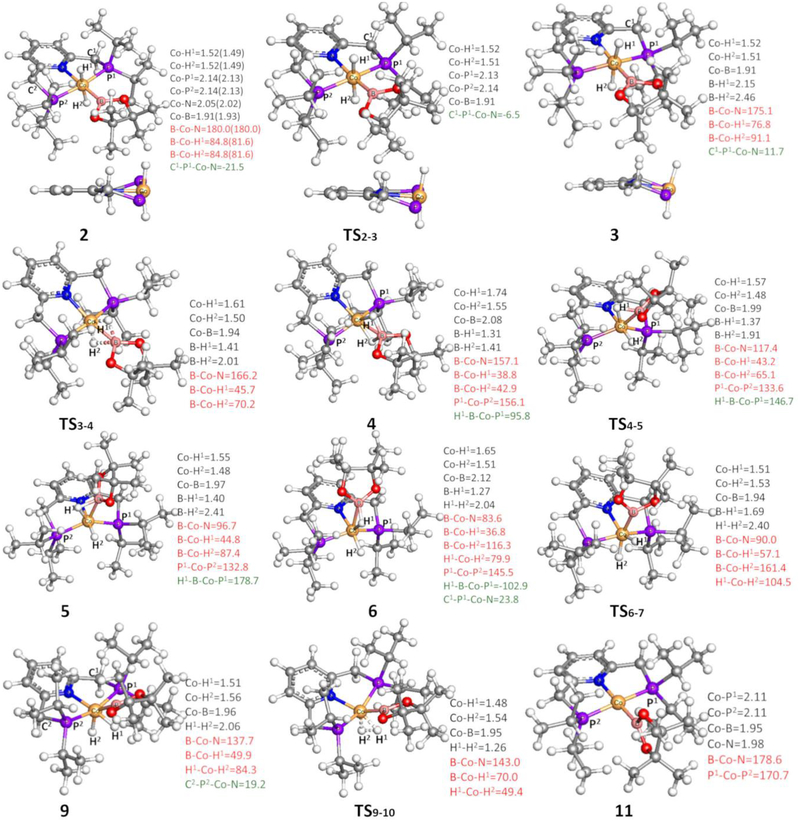
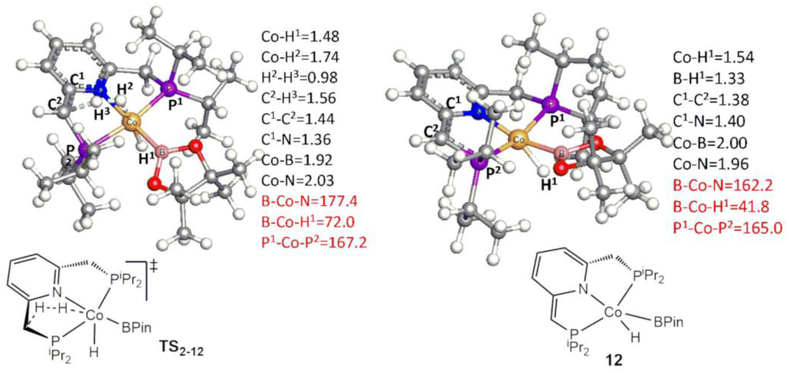
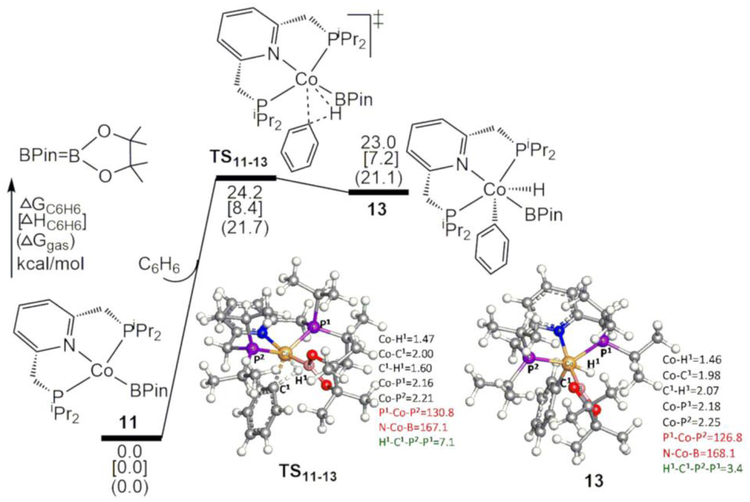
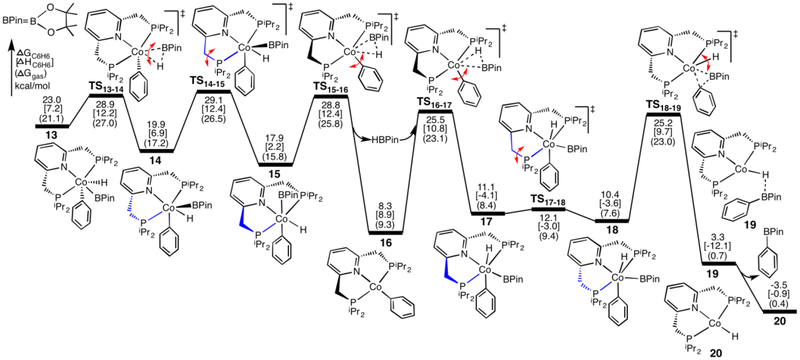
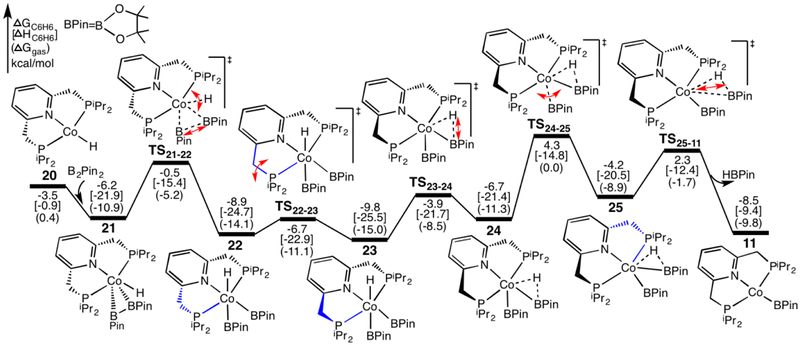
The mechanism for the borylation of an aromatic substrate by a cobalt pincer complex was investigated by density functional theory calculations. Experimental observations identified -(PNP)CoH(BPin) as the resting state in the borylation of five-membered heteroarenes, and 4-BPin-(PNP)Co(N)BPin as the resting state in the catalytic borylation of arene substrates. The active species, 4-R-(PNP)CoBPin (R=H, BPin), were generated by reductive elimination of H in the former, through Berry pseudorotation to the isomer, and N loss in the latter. The catalytic mechanism of the resulting Co(I) complex was computed to involve three main steps: C-H oxidative addition of the aromatic substrate (CH), reductive elimination of PhBPin, and regeneration of the active complex. The oxidative addition product formed through the most favorable pathway, where the breaking C-H bond of CH is parallel to a line between the two phosphine atoms, leaves the complex with a distorted PNP ligand, which rearranges to a more stable complex via dissociation and re-association of HBPin. Alternative pathways, σ-bond metathesis and the oxidative addition in which the breaking C-H bond is parallel to the Co-B bond, are predicted to be unlikely for this Co(I) complex. The thermodynamically favorable formation of the product PhBPin via reductive elimination drives the reaction forward. The active species regenerates through the oxidative addition of BPin and reductive elimination of HBPin. . Metal-ligand cooperation based on the ligand's aromatization/dearomatization, a common mechanism for heavy-metal pincer complexes, and the dissociation of one phosphine ligand, do not apply in this system. This study provides guidance for understanding important features of pincer ligands with first-transition-row metals that differ from those in heavier metal complexes.
通过密度泛函理论计算研究了钴钳形配合物对芳族底物进行硼化反应的机理。实验观察表明,在五元杂芳烃硼化反应中,-(PNP)CoH(BPin)为静止状态;在芳烃底物的催化硼化反应中,4-BPin-(PNP)Co(N)BPin为静止状态。活性物种4-R-(PNP)CoBPin(R = H,BPin)是通过前者中H的还原消除、经Berry假旋转转变为异构体以及后者中N的损失而生成的。计算得出,所得Co(I)配合物的催化机理涉及三个主要步骤:芳族底物的C-H氧化加成(CH)、PhBPin的还原消除以及活性配合物的再生。通过最有利途径形成的氧化加成产物,其中CH断裂的C-H键与两个膦原子之间的连线平行,使配合物带有扭曲的PNP配体,该配体通过HBPin的解离和重新缔合重排为更稳定的配合物。预测该Co(I)配合物不太可能通过σ键复分解以及断裂的C-H键与Co-B键平行的氧化加成等替代途径进行反应。通过还原消除形成产物PhBPin的热力学有利过程推动了反应的进行。活性物种通过BPin的氧化加成和HBPin的还原消除得以再生。基于配体芳构化/去芳构化的金属-配体协同作用(重金属钳形配合物的常见机理)以及一个膦配体的解离,在该体系中并不适用。本研究为理解第一过渡系金属钳形配体不同于重金属配合物的重要特征提供了指导。