Department of Orthodontics, Guiyang Hospital of Stomatology, Guiyang, China.
College of Stomatology, Chongqing Medical University, Chongqing, China.
J Cell Biochem. 2019 Jun;120(6):9277-9290. doi: 10.1002/jcb.28203. Epub 2019 Feb 5.
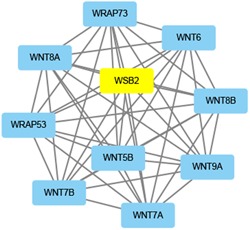
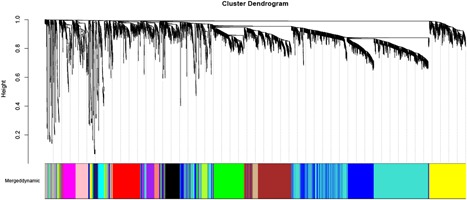
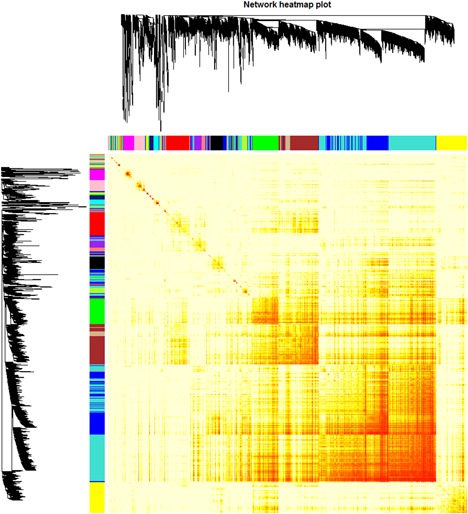
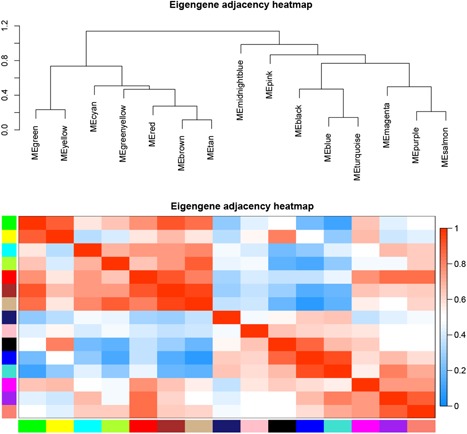
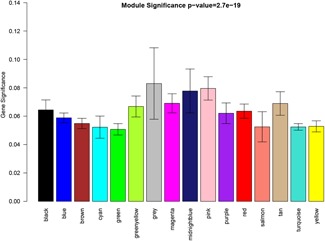
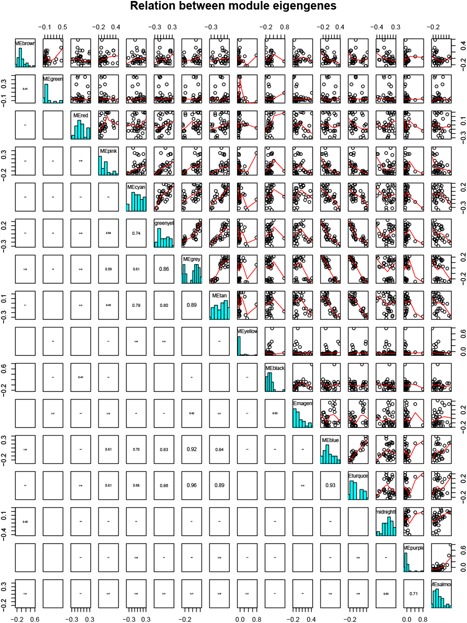
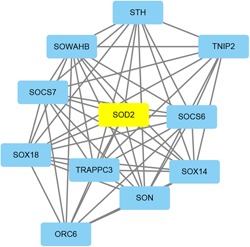
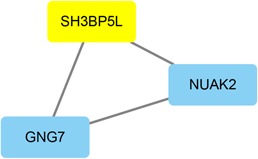
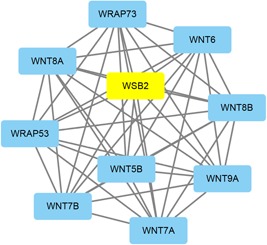
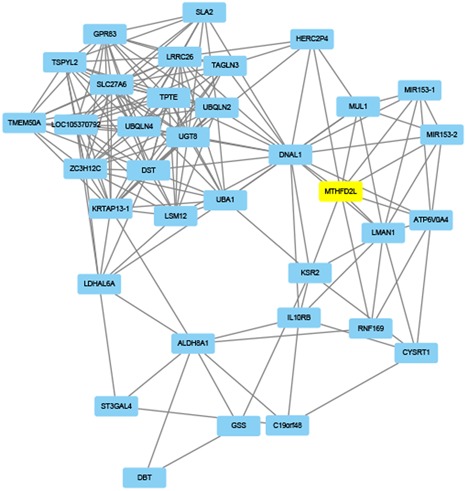
A growing number of studies provide epidemiological evidence linking obstructive sleep apnea (OSA) with a number of chronic disorders. Transcriptional analyses have been conducted to analyze the gene expression data. However, the weighted gene coexpression network analysis (WGCNA) method has not been applied to determine the transcriptional consequence of continuous positive airway pressure (CPAP) therapy in patients with severe OSA. The aim of this study was to identify key pathways and genes in patients with OSA that are influenced by CPAP treatment and uncover/unveil potential molecular mechanisms using WGCNA. We analyzed the microarray data of OSA (GSE 49800) listed in the Gene Expression Omnibus database. Coexpression modules were constructed using WGCNA. In addition, Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analysis were also conducted. After the initial data processing, 5101 expressed gene profiles were identified. Next, a weighted gene coexpression network was established and 16 modules of coexpressed genes were identified. The interaction analysis demonstrated a relative independence of gene expression in these modules. The black module, tan module, midnightblue module, pink module, and greenyellow module were significantly associated with the alterations in circulating leukocyte gene expression at baseline and after exposure to CPAP. The five hub genes were considered to be candidate OSA-related genes after CPAP treatment. Functional enrichment analysis revealed that steroid biosynthesis, amino sugar and nucleotide sugar metabolism, protein processing in the endoplasmic reticulum, and the insulin signaling pathway play critical roles in the development of OSA in circulating leukocyte gene expression at baseline and after exposure to CPAP. Using this new systems biology approach, we identified several genes and pathways that appear to be critical to OSA after CPAP treatment, and these findings provide a better understanding of OSA pathogenesis.
越来越多的研究提供了流行病学证据,将阻塞性睡眠呼吸暂停(OSA)与许多慢性疾病联系起来。已经进行了转录分析以分析基因表达数据。然而,加权基因共表达网络分析(WGCNA)方法尚未应用于确定严重 OSA 患者持续气道正压通气(CPAP)治疗的转录后果。本研究旨在使用 WGCNA 确定 CPAP 治疗影响 OSA 患者的关键途径和基因,并揭示/揭示潜在的分子机制。我们分析了基因表达综合数据库(GEO)中列出的 OSA 的微阵列数据(GSE49800)。使用 WGCNA 构建共表达模块。此外,还进行了基因本体论和京都基因与基因组百科全书富集分析。经过初步的数据处理,确定了 5101 个表达基因谱。接下来,建立了一个加权基因共表达网络,并确定了 16 个共表达基因模块。相互作用分析表明,这些模块中的基因表达具有相对独立性。黑色模块、棕褐色模块、午夜蓝色模块、粉红色模块和黄绿色模块与基线和暴露于 CPAP 后循环白细胞基因表达的变化显著相关。五个枢纽基因被认为是 CPAP 治疗后候选的 OSA 相关基因。功能富集分析表明,类固醇生物合成、氨基糖和核苷酸糖代谢、内质网蛋白加工以及胰岛素信号通路在基线和暴露于 CPAP 后循环白细胞基因表达的 OSA 发展中起着关键作用。使用这种新的系统生物学方法,我们确定了几个基因和途径,这些基因和途径似乎在 CPAP 治疗后的 OSA 中至关重要,这些发现为 OSA 的发病机制提供了更好的理解。