Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Nucleic Acids Res. 2019 Apr 23;47(7):e39. doi: 10.1093/nar/gkz068.
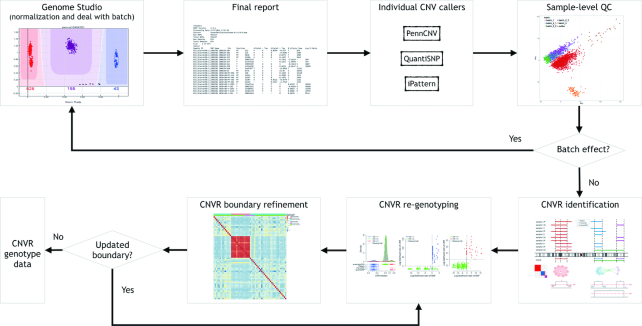
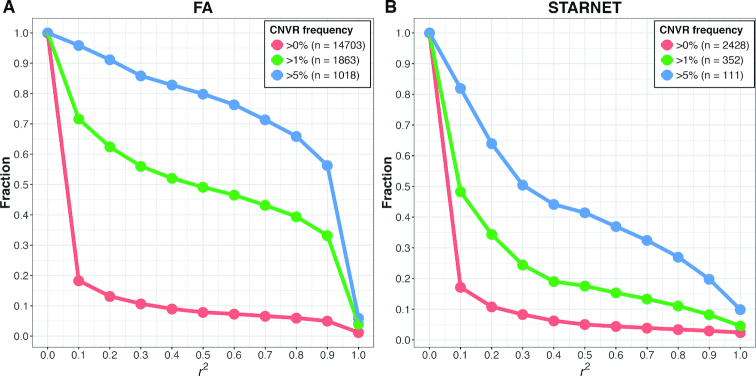
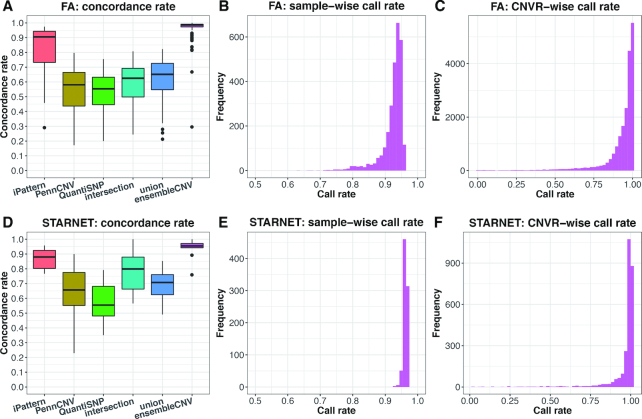
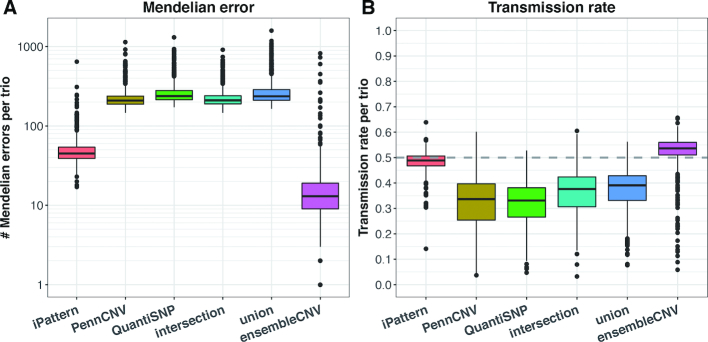
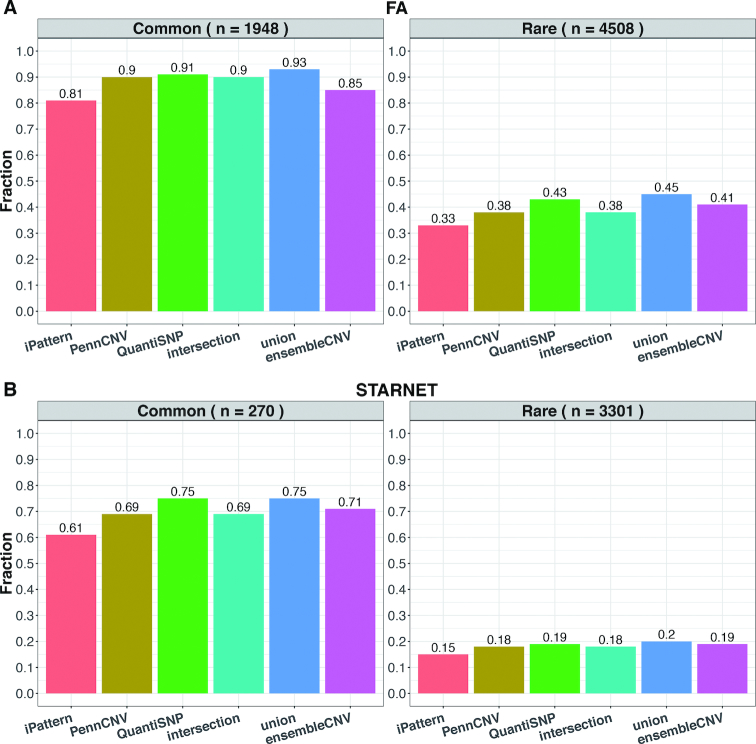
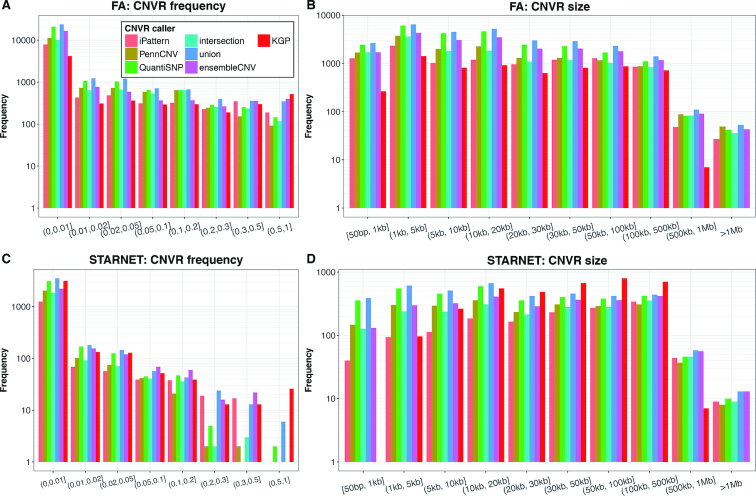
The associations between diseases/traits and copy number variants (CNVs) have not been systematically investigated in genome-wide association studies (GWASs), primarily due to a lack of robust and accurate tools for CNV genotyping. Herein, we propose a novel ensemble learning framework, ensembleCNV, to detect and genotype CNVs using single nucleotide polymorphism (SNP) array data. EnsembleCNV (a) identifies and eliminates batch effects at raw data level; (b) assembles individual CNV calls into CNV regions (CNVRs) from multiple existing callers with complementary strengths by a heuristic algorithm; (c) re-genotypes each CNVR with local likelihood model adjusted by global information across multiple CNVRs; (d) refines CNVR boundaries by local correlation structure in copy number intensities; (e) provides direct CNV genotyping accompanied with confidence score, directly accessible for downstream quality control and association analysis. Benchmarked on two large datasets, ensembleCNV outperformed competing methods and achieved a high call rate (93.3%) and reproducibility (98.6%), while concurrently achieving high sensitivity by capturing 85% of common CNVs documented in the 1000 Genomes Project. Given this CNV call rate and accuracy, which are comparable to SNP genotyping, we suggest ensembleCNV holds significant promise for performing genome-wide CNV association studies and investigating how CNVs predispose to human diseases.
疾病/特征与拷贝数变异(CNV)之间的关联尚未在全基因组关联研究(GWAS)中进行系统研究,主要是因为缺乏用于 CNV 基因分型的强大而准确的工具。在此,我们提出了一种新的集成学习框架,ensembleCNV,用于使用单核苷酸多态性(SNP)阵列数据检测和基因分型 CNV。ensembleCNV(a)在原始数据级别识别和消除批次效应;(b)通过启发式算法将来自多个现有调用者的个体 CNV 调用组装成 CNV 区域(CNVR),这些调用者具有互补的优势;(c)使用跨多个 CNVR 调整的全局信息重新对每个 CNVR 进行局部似然模型基因分型;(d)通过拷贝数强度中的局部相关结构细化 CNVR 边界;(e)提供直接的 CNV 基因分型,同时提供置信度评分,可直接用于下游质量控制和关联分析。在两个大型数据集上进行基准测试,ensembleCNV 优于竞争方法,实现了高调用率(93.3%)和可重复性(98.6%),同时通过捕获 1000 基因组计划中记录的 85%常见 CNV 实现了高灵敏度。鉴于这种 CNV 调用率和准确性与 SNP 基因分型相当,我们建议 ensembleCNV 在进行全基因组 CNV 关联研究和研究 CNV 如何导致人类疾病方面具有很大的潜力。