Chang Jia-Ming, Floden Evan W, Herrero Javier, Gascuel Olivier, Di Tommaso Paolo, Notredame Cedric
European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Cambridge CB10 1SD, UK.
Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Barcelona 08003, Spain.
Bioinformatics. 2021 Jul 12;37(11):1506-1514. doi: 10.1093/bioinformatics/btz082.
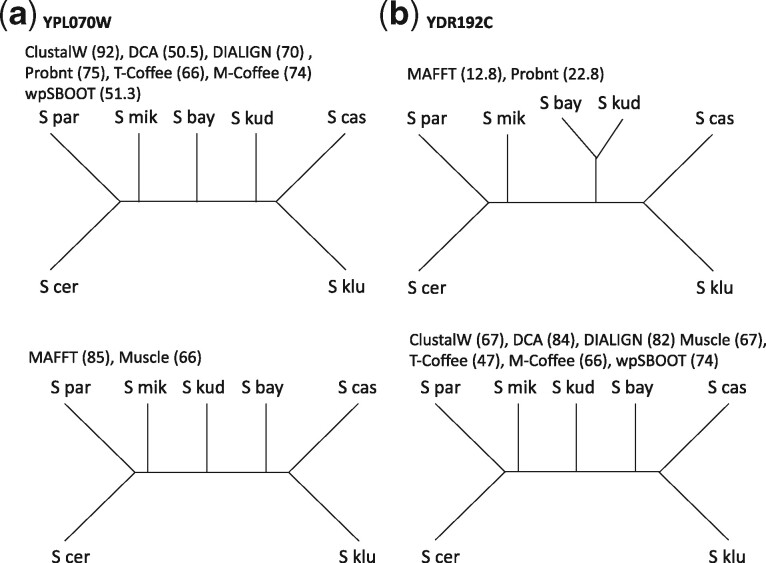
Most evolutionary analyses are based on pre-estimated multiple sequence alignment. Wong et al. established the existence of an uncertainty induced by multiple sequence alignment when reconstructing phylogenies. They were able to show that in many cases different aligners produce different phylogenies, with no simple objective criterion sufficient to distinguish among these alternatives.
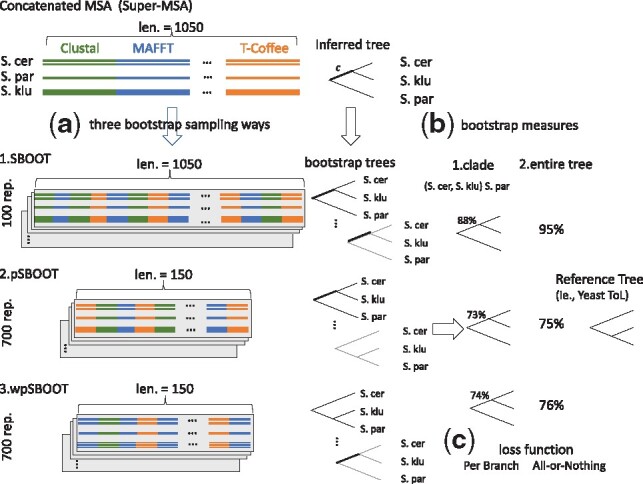
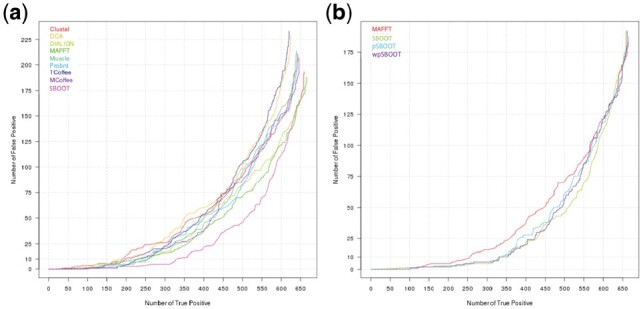
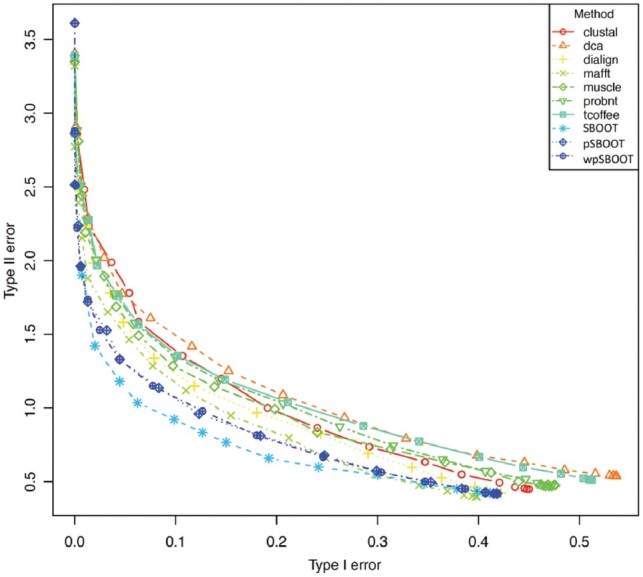
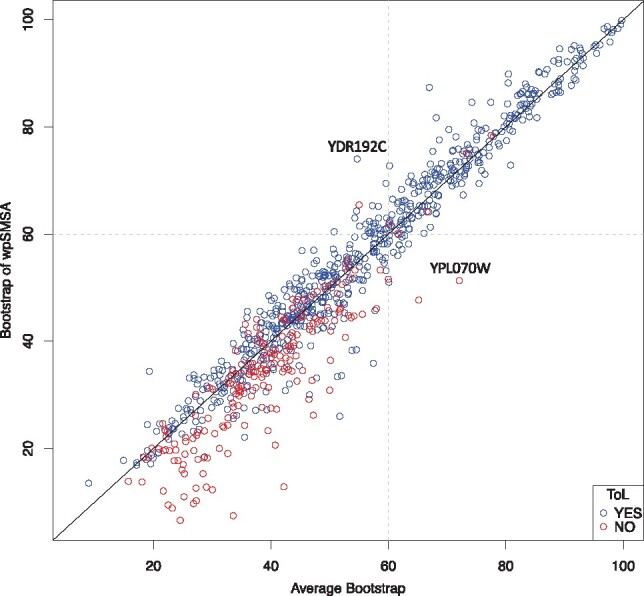
We demonstrate that incorporating MSA induced uncertainty into bootstrap sampling can significantly increase correlation between clade correctness and its corresponding bootstrap value. Our procedure involves concatenating several alternative multiple sequence alignments of the same sequences, produced using different commonly used aligners. We then draw bootstrap replicates while favoring columns of the more unique aligner among the concatenated aligners. We named this concatenation and bootstrapping method, Weighted Partial Super Bootstrap (wpSBOOT). We show on three simulated datasets of 16, 32 and 64 tips that our method improves the predictive power of bootstrap values. We also used as a benchmark an empirical collection of 853 one to one orthologous genes from seven yeast species and found wpSBOOT to significantly improve discrimination capacity between topologically correct and incorrect trees. Bootstrap values of wpSBOOT are comparable to similar readouts estimated using a single method. However, for reduced trees by 50 and 95% bootstrap thresholds, wpSBOOT comes out the lowest Type I error (less FP).
The automated generation of replicates has been implemented in the T-Coffee package, which is available as open source freeware available from www.tcoffee.org.
Supplementary data are available at Bioinformatics online.
大多数进化分析基于预先估计的多序列比对。Wong等人在重建系统发育树时证实了多序列比对会引发不确定性。他们发现,在许多情况下,不同的比对工具会产生不同的系统发育树,且没有简单的客观标准足以区分这些不同结果。
我们证明,将多序列比对引发的不确定性纳入自展抽样能够显著提高分支正确性与其相应自展值之间的相关性。我们的方法包括将使用不同常用比对工具生成的同一序列的多个替代多序列比对连接起来。然后在连接后的比对中更倾向于选择更独特的比对工具的列来进行自展重复抽样。我们将这种连接和自展方法命名为加权部分超级自展(wpSBOOT)。我们在三个分别包含16、32和64个末端的模拟数据集上表明,我们的方法提高了自展值的预测能力。我们还以来自七个酵母物种的853个一对一直系同源基因的实证集合作为基准,发现wpSBOOT显著提高了拓扑正确和不正确树之间的区分能力。wpSBOOT的自展值与使用单一方法估计的类似读数相当。然而,对于通过50%和95%自展阈值简化的树,wpSBOOT的I型错误率最低(假阳性更少)。
重复抽样的自动生成已在T-Coffee软件包中实现,该软件包可从www.tcoffee.org作为开源免费软件获取。
补充数据可在《生物信息学》在线获取。