Unité Bioinformatique Evolutive, C3BI USR 3756, Institut Pasteur & CNRS, Paris, France.
Hub Bioinformatique et Biostatistique, C3BI USR 3756, Institut Pasteur & CNRS, Paris, France.
Nature. 2018 Apr;556(7702):452-456. doi: 10.1038/s41586-018-0043-0. Epub 2018 Apr 18.
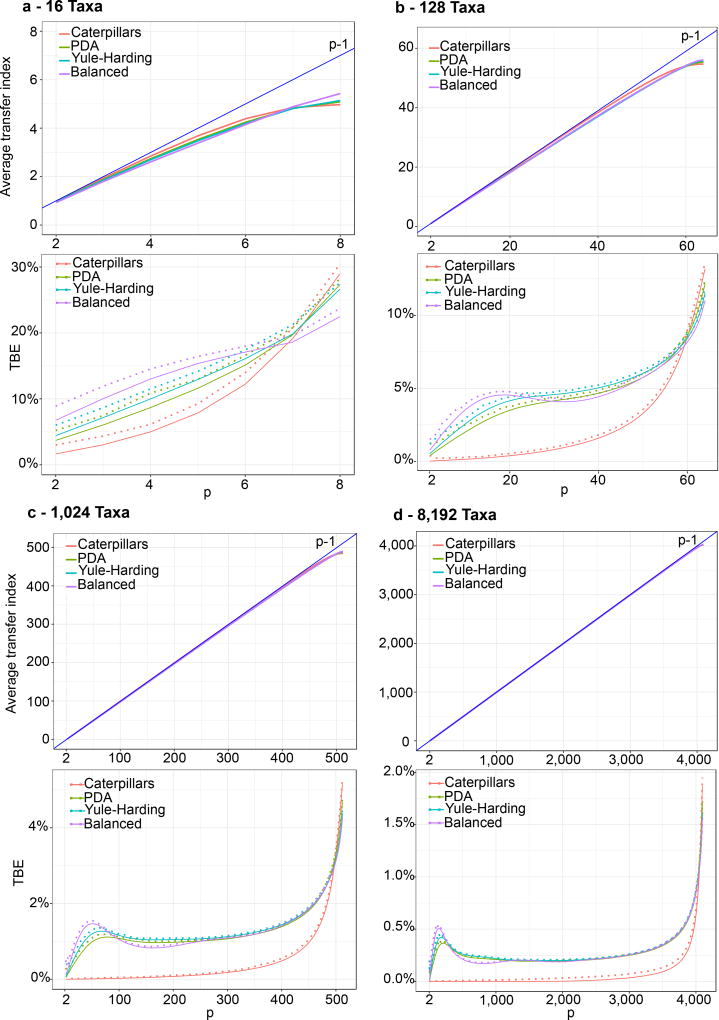
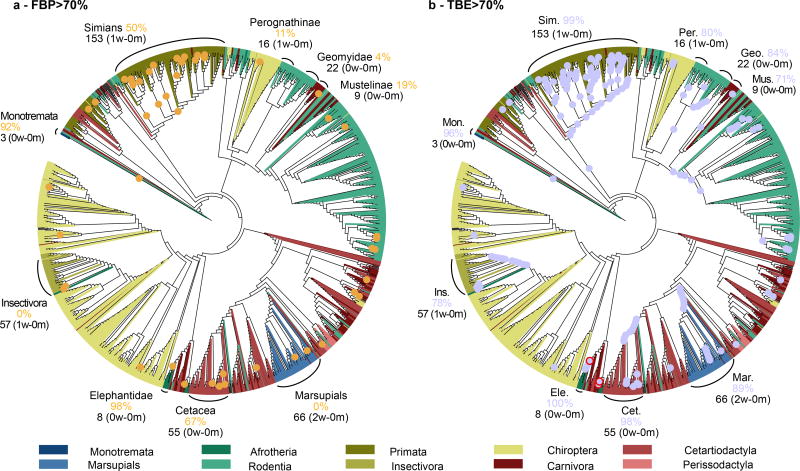
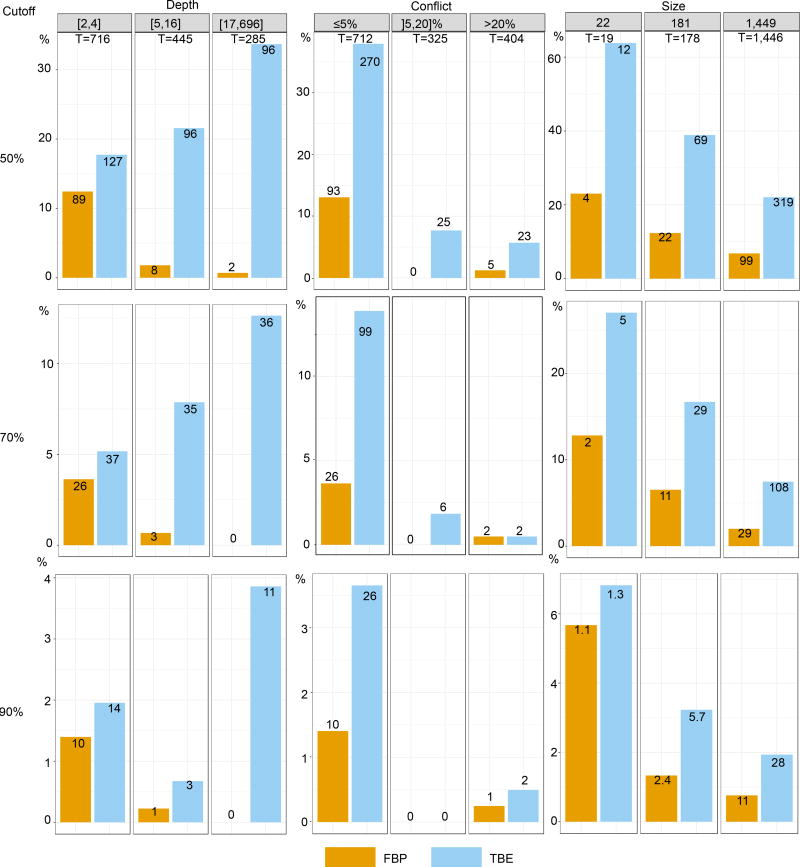
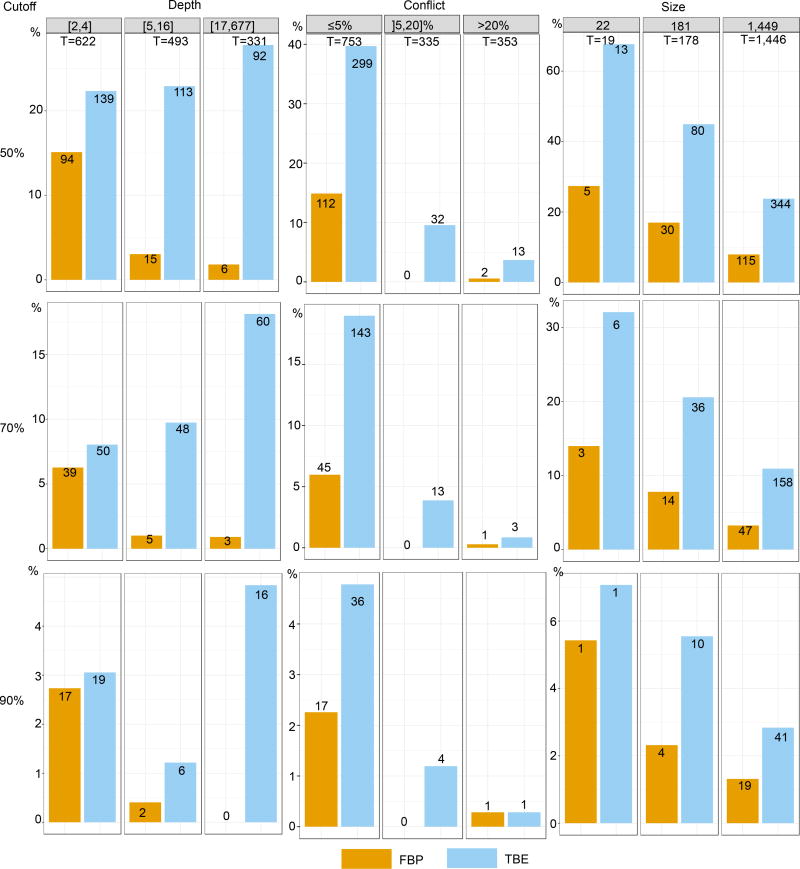
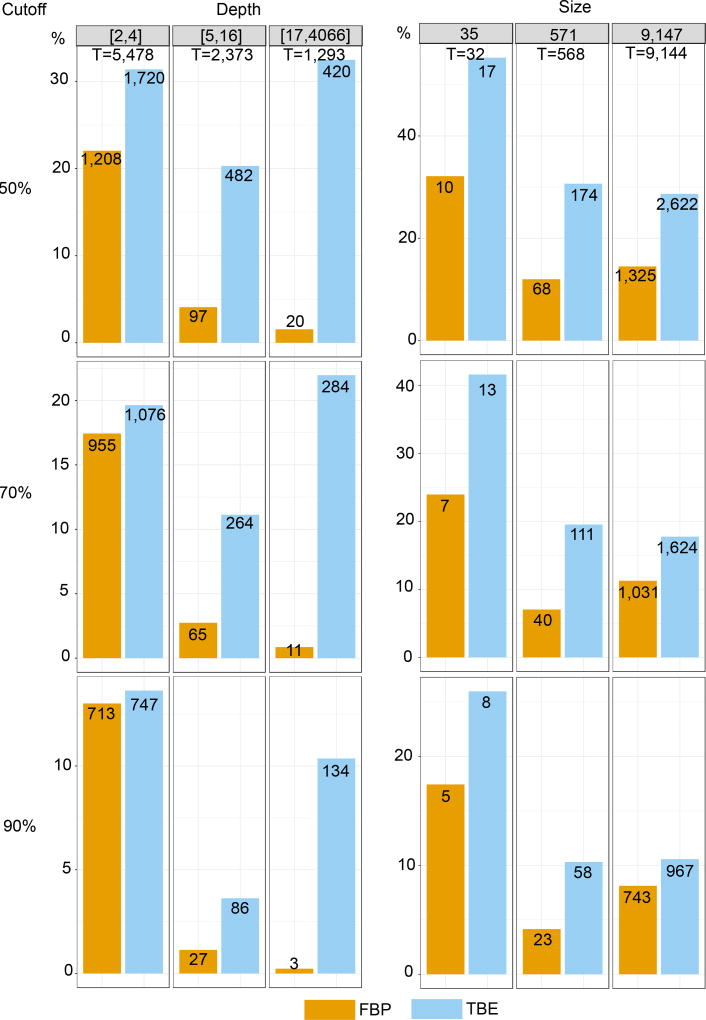
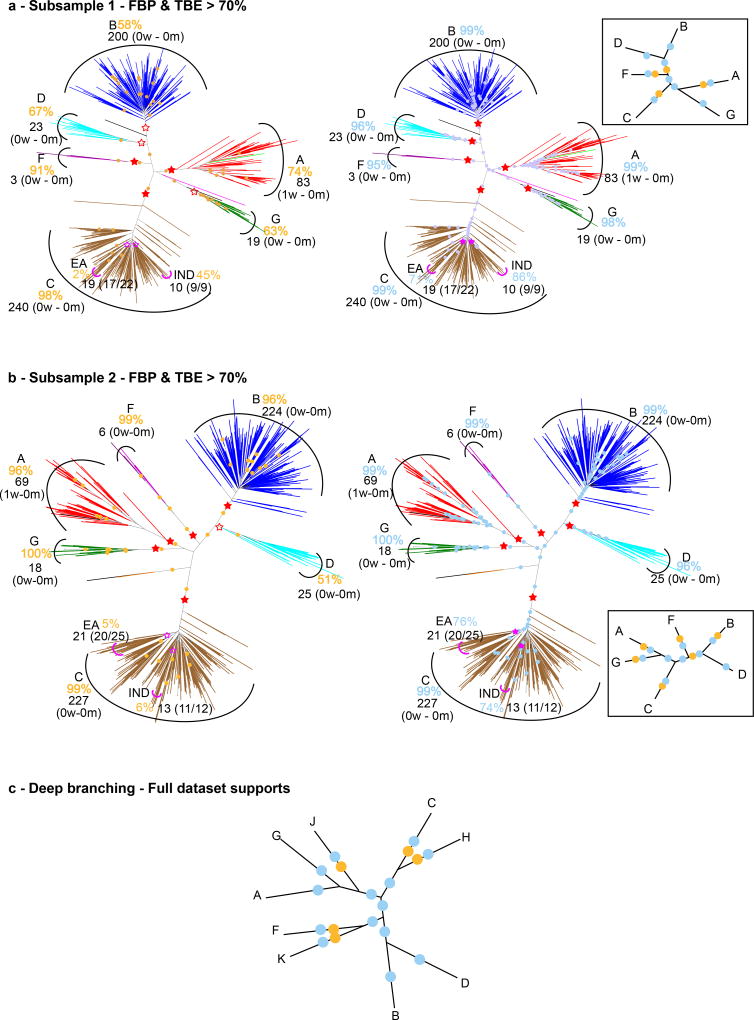
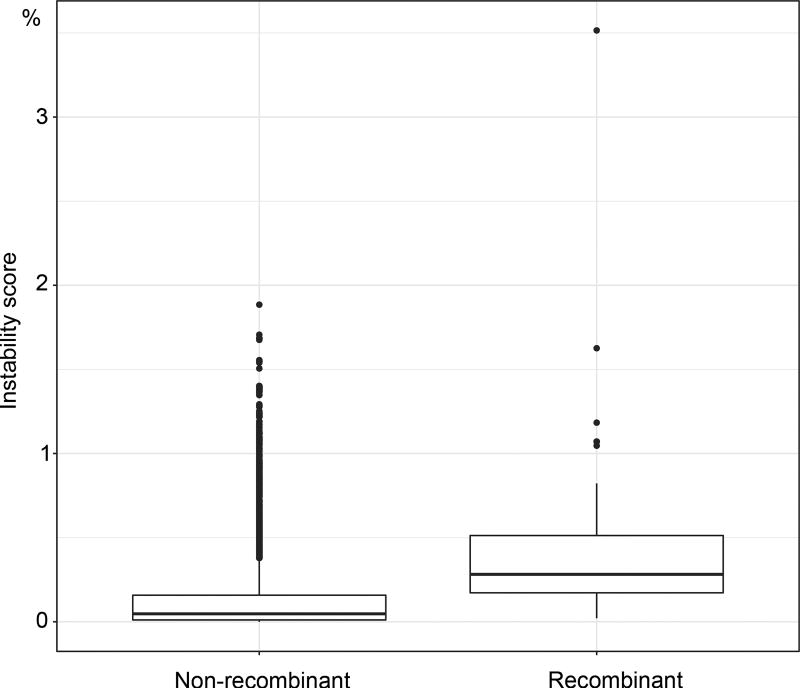
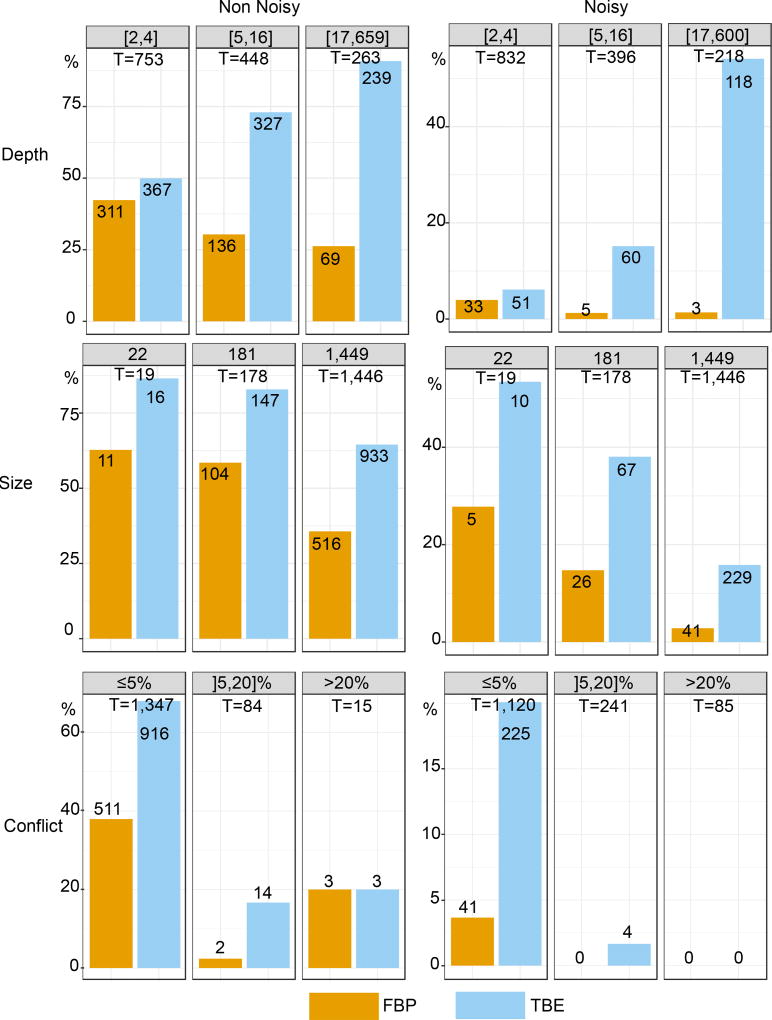
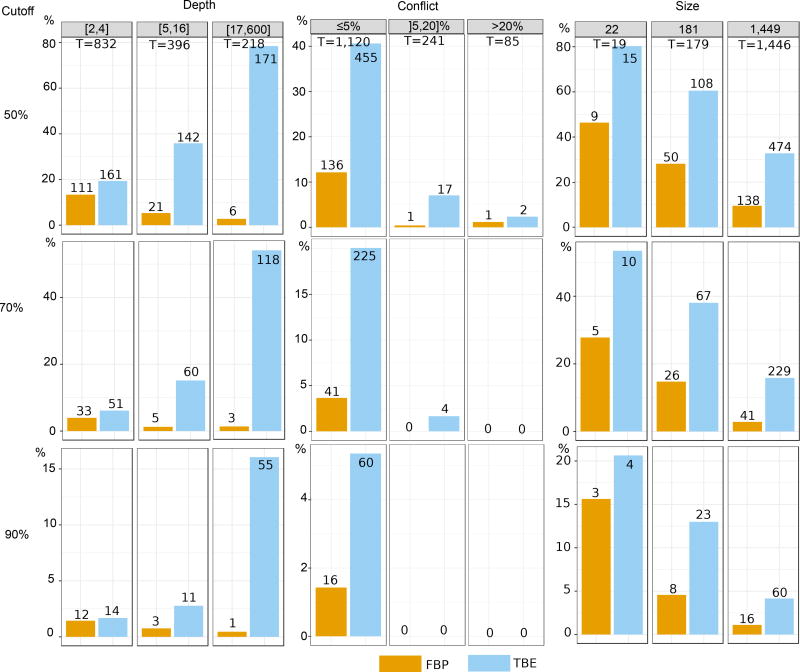
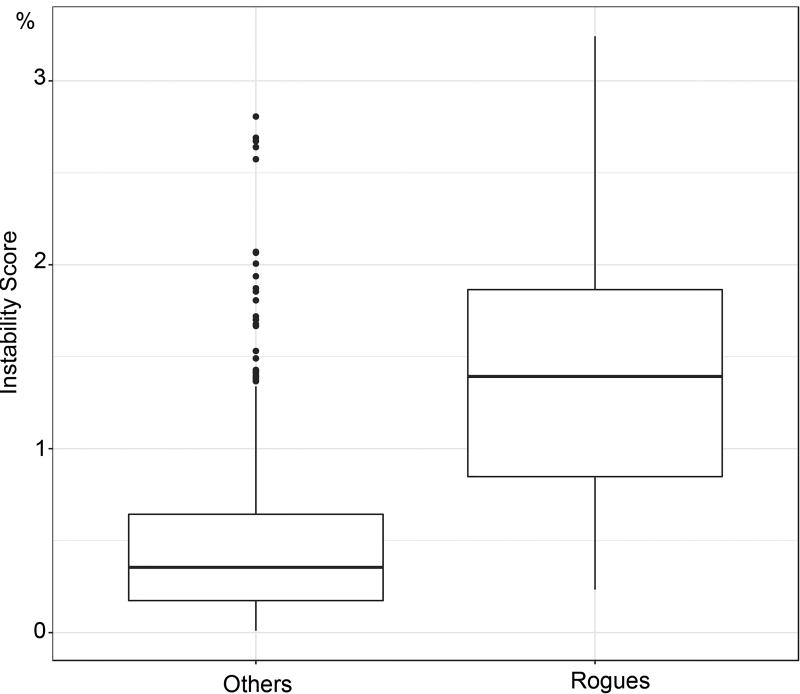
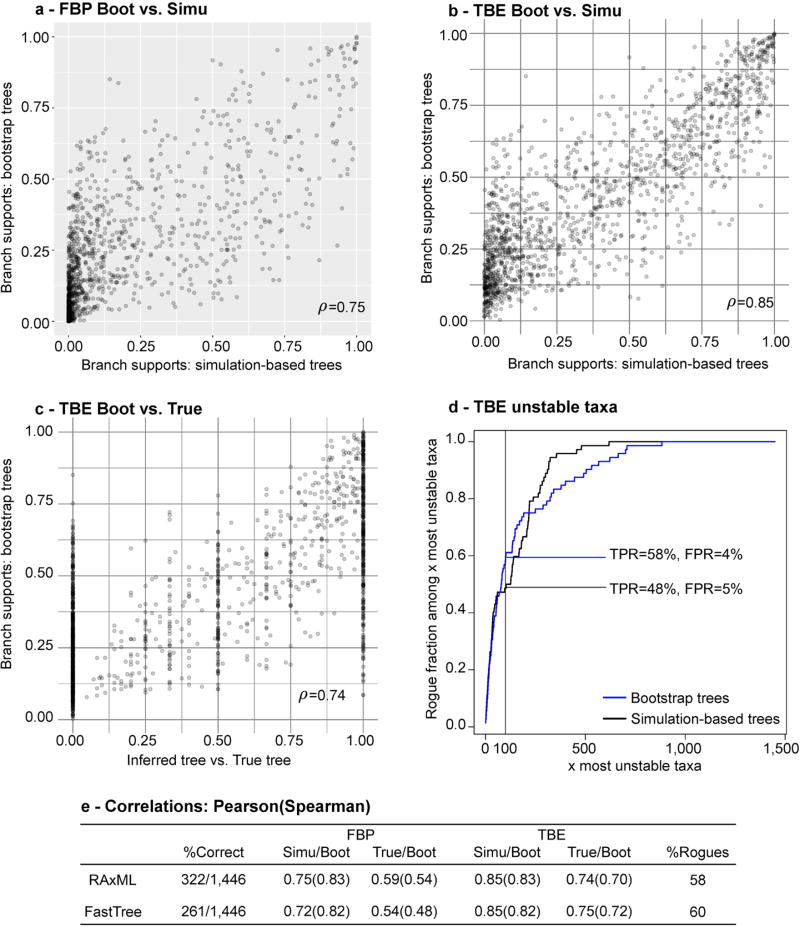
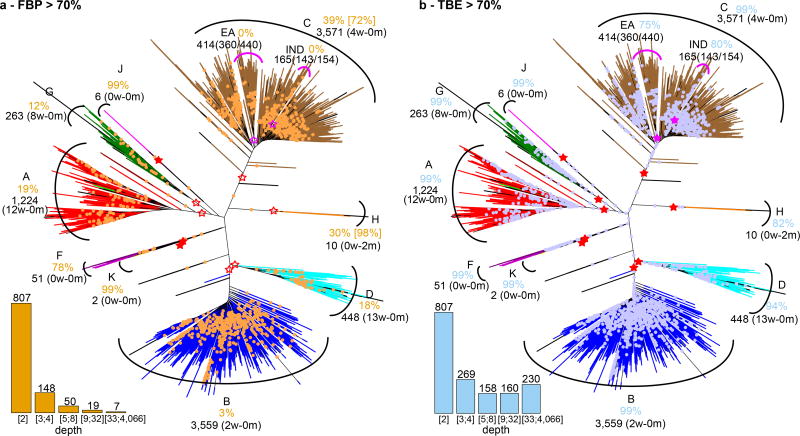
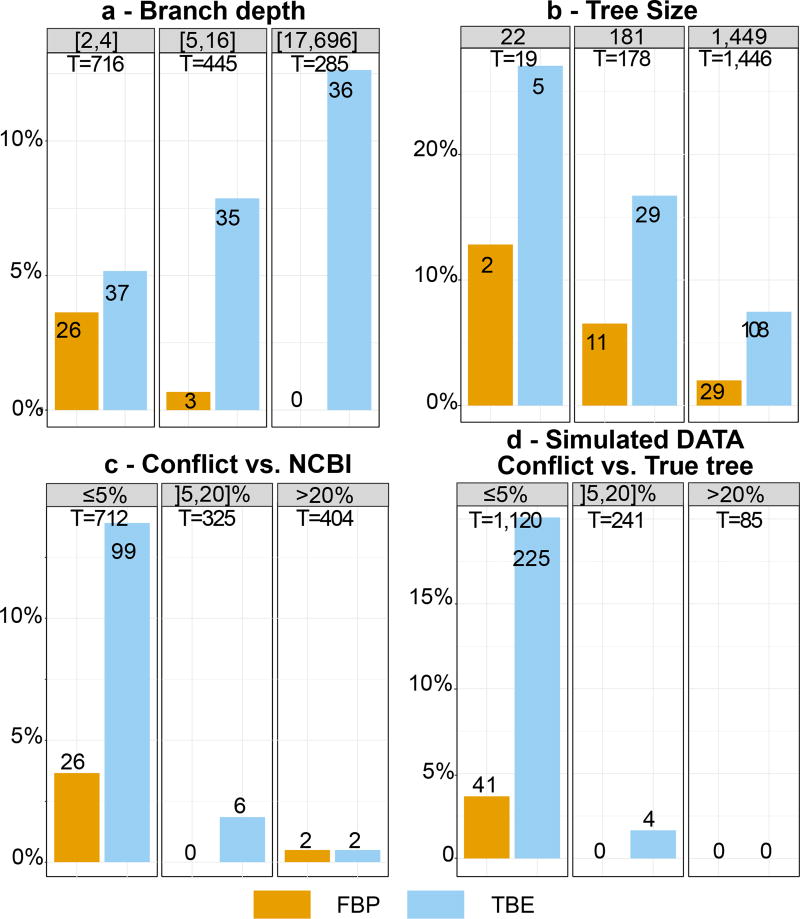
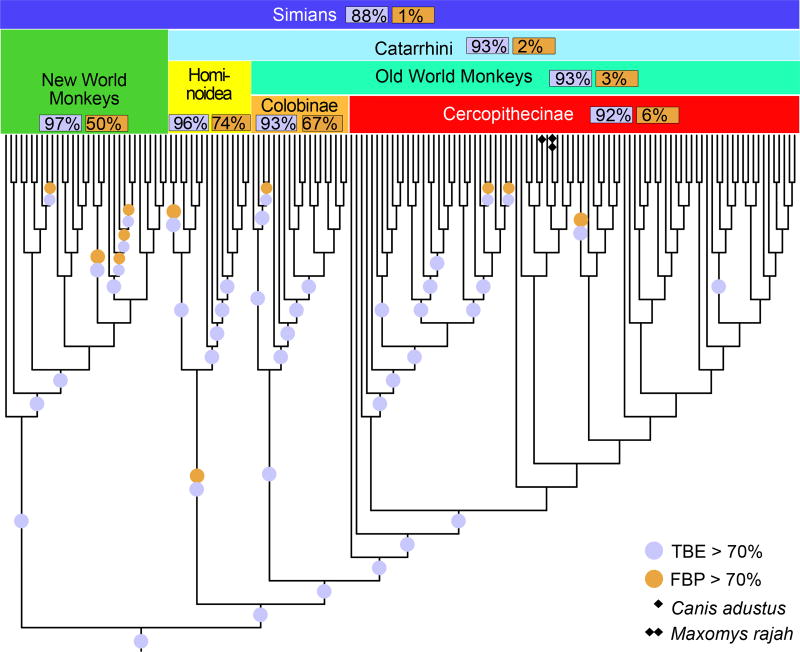
Felsenstein's application of the bootstrap method to evolutionary trees is one of the most cited scientific papers of all time. The bootstrap method, which is based on resampling and replications, is used extensively to assess the robustness of phylogenetic inferences. However, increasing numbers of sequences are now available for a wide variety of species, and phylogenies based on hundreds or thousands of taxa are becoming routine. With phylogenies of this size Felsenstein's bootstrap tends to yield very low supports, especially on deep branches. Here we propose a new version of the phylogenetic bootstrap in which the presence of inferred branches in replications is measured using a gradual 'transfer' distance rather than the binary presence or absence index used in Felsenstein's original version. The resulting supports are higher and do not induce falsely supported branches. The application of our method to large mammal, HIV and simulated datasets reveals their phylogenetic signals, whereas Felsenstein's bootstrap fails to do so.
费希尔斯坦因将自举法应用于进化树,是有史以来被引用最多的科学论文之一。自举法基于重采样和复制,被广泛用于评估系统发育推断的稳健性。然而,现在有越来越多的序列可用于各种物种,基于数百或数千分类群的系统发育已经成为常规。对于这种大小的系统发育,费希尔斯坦因的自举法往往会产生非常低的支持率,尤其是在深分支上。在这里,我们提出了一种新的系统发育自举法版本,其中在复制中推断分支的存在使用逐渐的“转移”距离来衡量,而不是费希尔斯坦因原始版本中使用的二元存在或不存在索引。由此产生的支持率更高,并且不会产生虚假支持的分支。我们的方法在大型哺乳动物、HIV 和模拟数据集上的应用揭示了它们的系统发育信号,而费希尔斯坦因的自举法却无法做到这一点。