Computational Biology of Infection Research, Helmholtz Centre for Infection Research, Braunschweig, 38124, Germany.
Formerly Department of Algorithmic Bioinformatics, Heinrich-Heine University Düsseldorf, Düsseldorf, 40225, Germany.
Microbiome. 2019 Feb 8;7(1):17. doi: 10.1186/s40168-019-0633-6.
Shotgun metagenome data sets of microbial communities are highly diverse, not only due to the natural variation of the underlying biological systems, but also due to differences in laboratory protocols, replicate numbers, and sequencing technologies. Accordingly, to effectively assess the performance of metagenomic analysis software, a wide range of benchmark data sets are required.
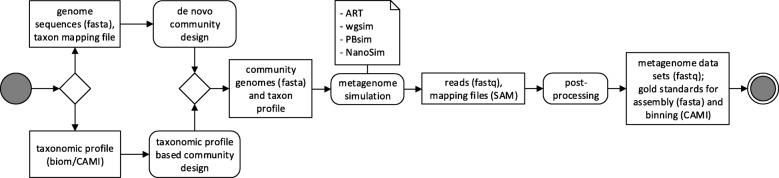
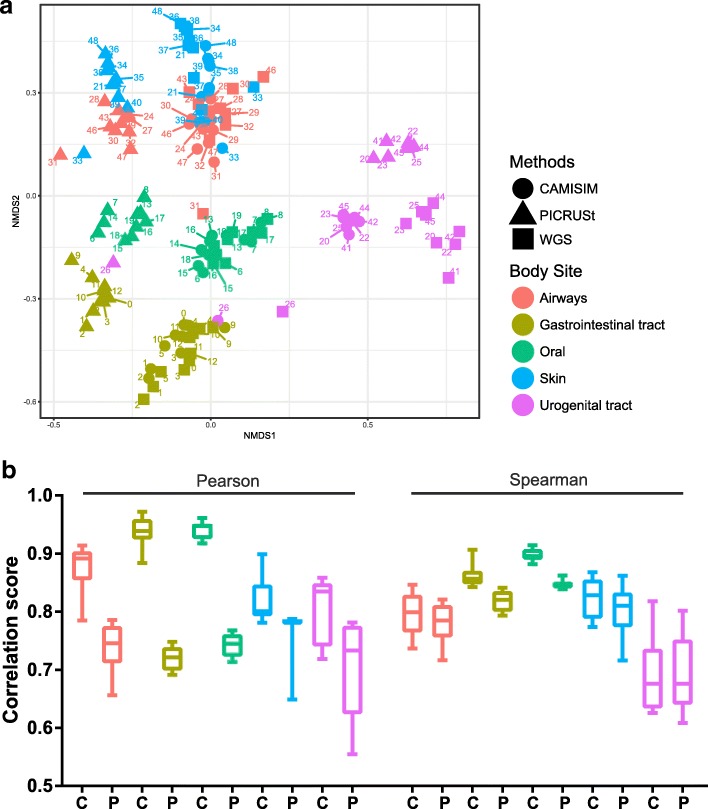
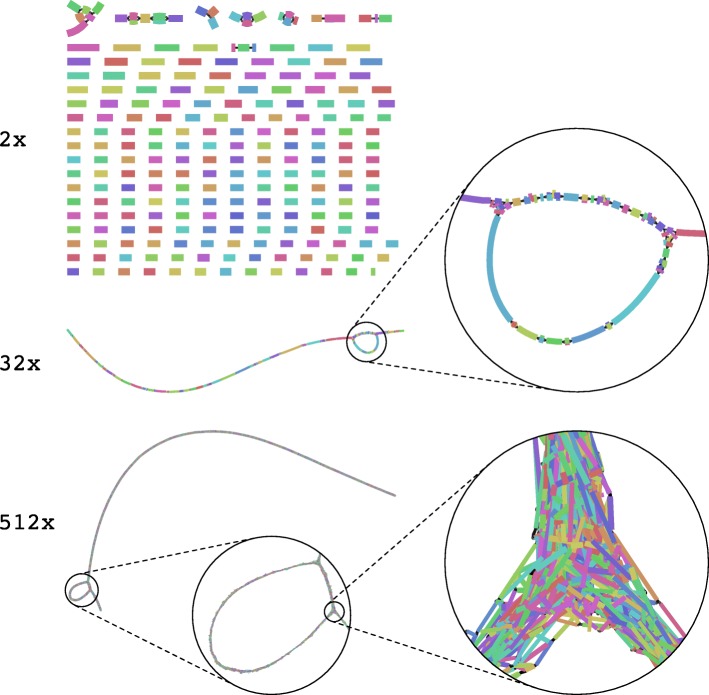
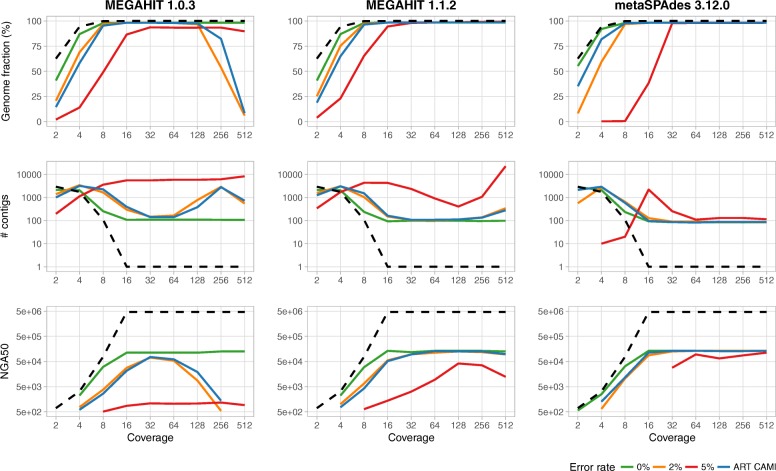
We describe the CAMISIM microbial community and metagenome simulator. The software can model different microbial abundance profiles, multi-sample time series, and differential abundance studies, includes real and simulated strain-level diversity, and generates second- and third-generation sequencing data from taxonomic profiles or de novo. Gold standards are created for sequence assembly, genome binning, taxonomic binning, and taxonomic profiling. CAMSIM generated the benchmark data sets of the first CAMI challenge. For two simulated multi-sample data sets of the human and mouse gut microbiomes, we observed high functional congruence to the real data. As further applications, we investigated the effect of varying evolutionary genome divergence, sequencing depth, and read error profiles on two popular metagenome assemblers, MEGAHIT, and metaSPAdes, on several thousand small data sets generated with CAMISIM.
CAMISIM can simulate a wide variety of microbial communities and metagenome data sets together with standards of truth for method evaluation. All data sets and the software are freely available at https://github.com/CAMI-challenge/CAMISIM.
微生物群落的 shotgun 宏基因组数据集高度多样化,不仅由于基础生物系统的自然变化,还由于实验室方案、重复数量和测序技术的差异。因此,为了有效地评估宏基因组分析软件的性能,需要广泛的基准数据集。
我们描述了 CAMISIM 微生物群落和宏基因组模拟器。该软件可以模拟不同的微生物丰度分布、多样本时间序列和差异丰度研究,包括真实和模拟的菌株水平多样性,并根据分类分布或从头生成二级和三级测序数据。为序列组装、基因组分类、分类分类和分类分析创建了黄金标准。CAMSIM 生成了第一届 CAMI 挑战赛的基准数据集。对于人类和小鼠肠道微生物组的两个模拟多样本数据集,我们观察到与真实数据高度的功能一致性。作为进一步的应用,我们研究了在数千个使用 CAMISIM 生成的小数据集上,变化的进化基因组差异、测序深度和读取错误分布对两个流行的宏基因组组装器 MEGAHIT 和 metaSPAdes 的影响。
CAMISIM 可以模拟各种微生物群落和宏基因组数据集,并为方法评估提供真实标准。所有数据集和软件均可在 https://github.com/CAMI-challenge/CAMISIM 上免费获得。