Medical Big Data and Bioinformatics Research Centre, First Affiliated Hospital of Gannan Medical University, Ganzhou, 341000, Jiangxi, China.
Department of Bioinformatics, Fujian Key Laboratory of Medical Bioinformatics, Key Laboratory of Ministry of Education for Gastrointestinal Cancer, Fujian Medical University, Fuzhou, 350122, Fujian, China.
BMC Genomics. 2019 Feb 13;20(1):134. doi: 10.1186/s12864-019-5502-y.
The amount of RNA per cell, namely the transcriptome size, may vary under many biological conditions including tumor. If the transcriptome size of two cells is different, direct comparison of the expression measurements on the same amount of total RNA for two samples can only identify genes with changes in the relative mRNA abundances, i.e., cellular mRNA concentration, rather than genes with changes in the absolute mRNA abundances.
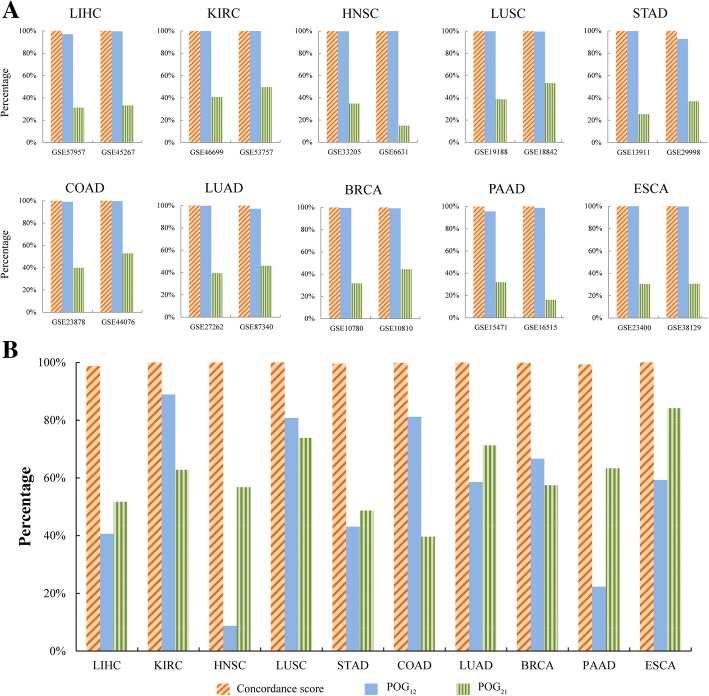
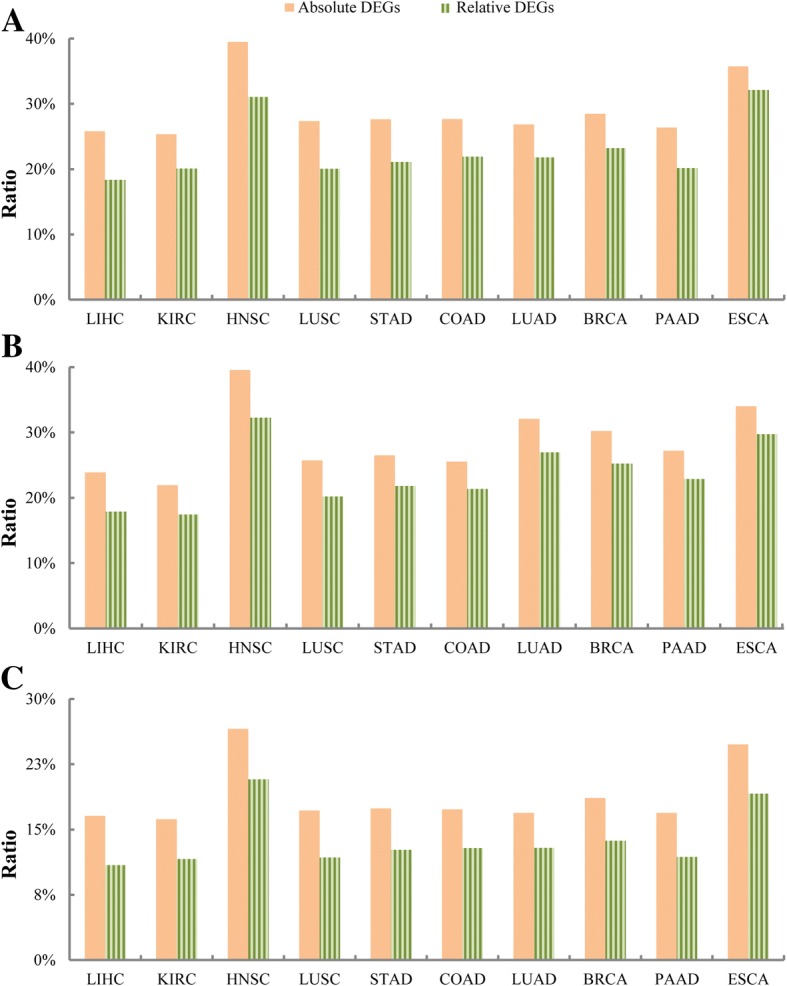
Our recently proposed RankCompV2 algorithm identify differentially expressed genes (DEGs) through comparing the relative expression orderings (REOs) of disease samples with that of normal samples. We reasoned that both the mRNA concentration and the absolute abundances of these DEGs must have changes in disease samples. In simulation experiments, this method showed excellent performance for identifying DEGs between normal and disease samples with different transcriptome sizes. Through analyzing data for ten cancer types, we found that a significantly higher proportion of the DEGs with absolute mRNA abundance changes overlapped or directly interacted with known cancer driver genes and anti-cancer drug targets than that of the DEGs only with mRNA concentration changes alone identified by the traditional methods. The DEGs with increased absolute mRNA abundances were enriched in DNA damage-related pathways, while DEGs with decreased absolute mRNA abundances were enriched in immune and metabolism associated pathways.
Both the mRNA concentration and the absolute abundances of the DEGs identified through REOs comparison change in disease samples in comparison with normal samples. In cancers these genes might play more important upstream roles in carcinogenesis.
细胞中的 RNA 量,即转录组大小,可能在许多生物学条件下发生变化,包括肿瘤。如果两个细胞的转录组大小不同,那么直接比较两个样本中相同量的总 RNA 的表达测量值,只能识别相对 mRNA 丰度(即细胞内 mRNA 浓度)发生变化的基因,而不能识别绝对 mRNA 丰度发生变化的基因。
我们最近提出的 RankCompV2 算法通过比较疾病样本与正常样本的相对表达排序(REO)来识别差异表达基因(DEGs)。我们推断,这些 DEGs 的 mRNA 浓度和绝对丰度都必须在疾病样本中发生变化。在模拟实验中,该方法在识别不同转录组大小的正常和疾病样本之间的 DEGs 方面表现出优异的性能。通过分析十种癌症类型的数据,我们发现,与传统方法仅通过 mRNA 浓度变化识别的 DEGs 相比,具有绝对 mRNA 丰度变化的 DEGs 与已知的癌症驱动基因和抗癌药物靶点的重叠或直接相互作用的比例显著更高。具有增加的绝对 mRNA 丰度的 DEGs 富集在与 DNA 损伤相关的途径中,而具有降低的绝对 mRNA 丰度的 DEGs 富集在免疫和代谢相关途径中。
与正常样本相比,通过 REO 比较识别的 DEGs 的 mRNA 浓度和绝对丰度在疾病样本中都发生了变化。在癌症中,这些基因可能在致癌作用中发挥更重要的上游作用。