Department of informatics, University of Oslo, Oslo, Norway.
Department of Mathematics, University of Oslo, Oslo, Norway.
PLoS Comput Biol. 2019 Feb 19;15(2):e1006731. doi: 10.1371/journal.pcbi.1006731. eCollection 2019 Feb.
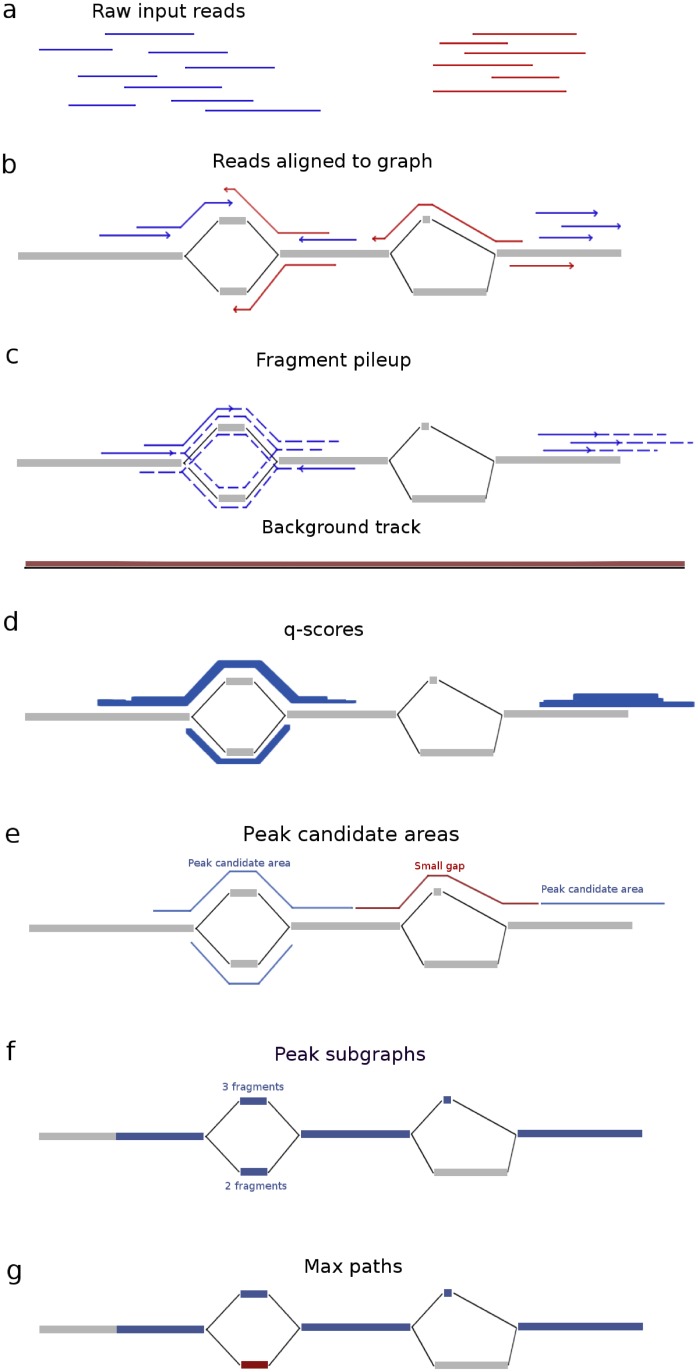
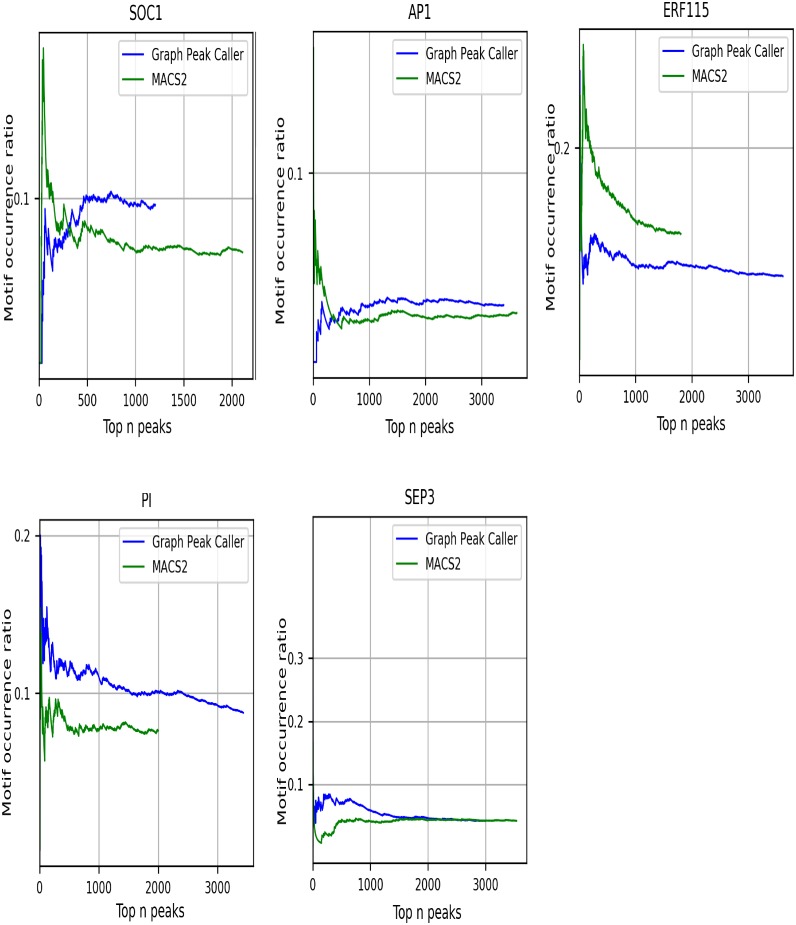
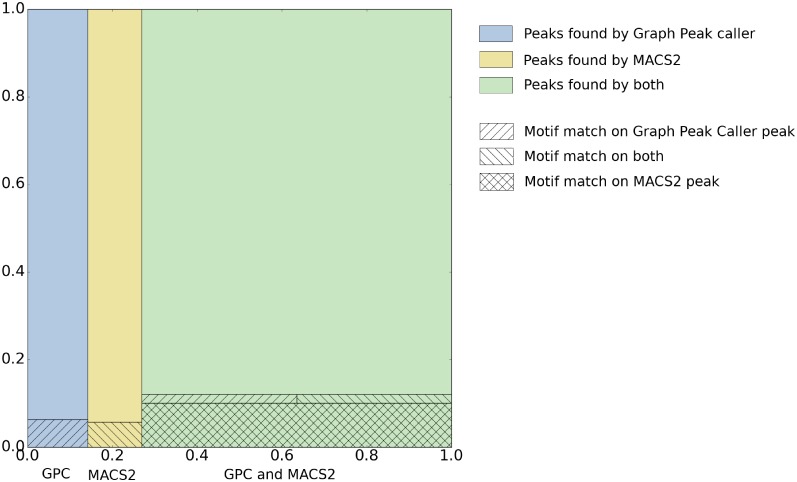
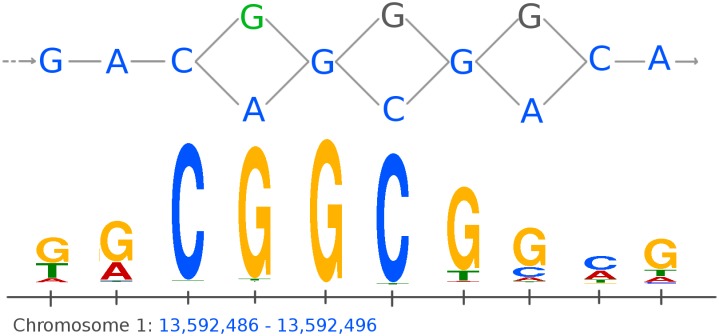
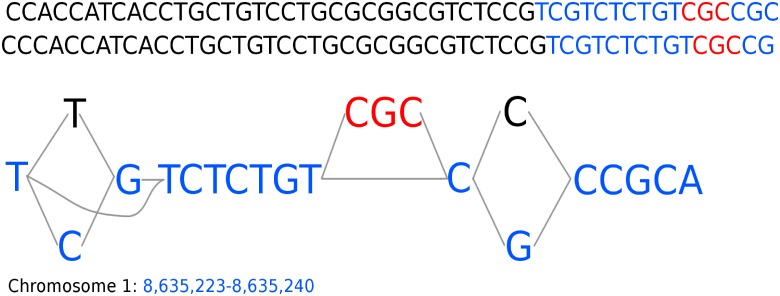
Graph-based representations are considered to be the future for reference genomes, as they allow integrated representation of the steadily increasing data on individual variation. Currently available tools allow de novo assembly of graph-based reference genomes, alignment of new read sets to the graph representation as well as certain analyses like variant calling and haplotyping. We here present a first method for calling ChIP-Seq peaks on read data aligned to a graph-based reference genome. The method is a graph generalization of the peak caller MACS2, and is implemented in an open source tool, Graph Peak Caller. By using the existing tool vg to build a pan-genome of Arabidopsis thaliana, we validate our approach by showing that Graph Peak Caller with a pan-genome reference graph can trace variants within peaks that are not part of the linear reference genome, and find peaks that in general are more motif-enriched than those found by MACS2.
基于图的表示被认为是参考基因组的未来,因为它们允许对个体变异的不断增加的数据进行集成表示。目前可用的工具允许基于图的参考基因组的从头组装,将新的读取集与图表示对齐,以及某些分析,如变体调用和单倍型分析。我们在这里提出了一种在基于图的参考基因组上对齐的读取数据上调用 ChIP-Seq 峰的方法。该方法是 MACS2 的峰调用器的图概括,并且在开源工具 Graph Peak Caller 中实现。通过使用现有的工具 vg 构建拟南芥的泛基因组,我们通过显示 Graph Peak Caller 与泛基因组参考图可以跟踪不在线性参考基因组中的峰内的变体,并找到通常比 MACS2 发现的峰更富含基序的峰,从而验证了我们的方法。