An Jinxia, Yang Jie, Wang Yan, Wang Yanxia, Xu Baicheng, Xie Guangmei, Chai Sanming, Liu Xiaoling, Xu Sijuan, Wen Xiaoxiao, He Qing, Liu Huijun, Li Chen, Dey Subrata Kumar, Ni Yali, Banerjee Santasree
Gansu Provincial Maternity and Child-care Hospital, Lanzhou, China.
Lanzhou University Second Hospital, Lanzhou, China.
Front Genet. 2019 Feb 5;10:1. doi: 10.3389/fgene.2019.00001. eCollection 2019.
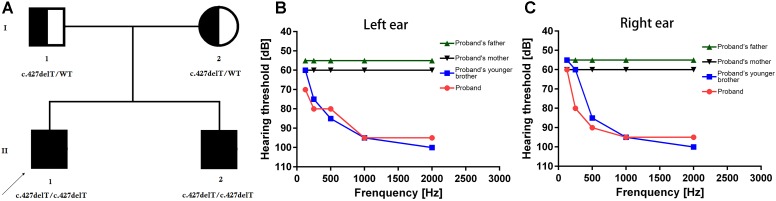
Hereditary hearing impairment is one of the major and common birth defects in Chinese population. Non-syndromic sensorineural hearing loss (NSHL) is the most common types of hereditary hearing impairment. Genotypically and phenotypically NSHL is extremely heterogenous and follow either autosomal dominant or autosomal recessive or X-linked mode of inheritance. Presently, 127 genes have been identified to be associated with both syndromic and (NSHL). Here, we studied a Chinese family with moderate and profound hearing impairment. The proband is a 30-year old Chinese man. The proband was born with normal hearing and at the age of 5-years, the proband was first noticed with hearing impairment. Gradually and progressively the proband was presented with loss of hearing in his both right and left ears at the age of 30 years. The clinical symptoms, age of onset or progression to loss of hearing was similar in both the proband and his younger brother. The proband's parents are phenotypically normal and non-consanguineous. Clinical diagnosis of the proband and his younger brother has been done by classical pure tone audiogram (PTA). Computed Tomography (CT) found no abnormality in bilateral external ear, middle ear and inner ear. Targeted next generation sequencing was performed with a panel of 127 genes reported to be associated with hereditary hearing impairment. A novel homozygous single nucleotide deletion (c.427delT) in exon 4 of gene has been identified in proband and in his younger brother. Sanger sequencing confirmed that proband's father and mother are carrying this mutation in a heterozygous manner. This mutation has not been identified in 100 normal healthy control individuals. This mutation (c.427delT) causes frameshift (p.Tyr143Ilefs19) which leads to the formation of a truncated ILDR1 protein of 162 amino acids instead of the wild type ILDR1 protein of 546 amino acids. associated hereditary hearing impairment is very rare and this is the first report of identifying a mutation in gene associated with hereditary hearing impairment in Chinese population. Our present study also emphasized the significance of rapid, accurate and cost-effective screening for the patient with hereditary hearing impairment by targeted next generation sequencing.
遗传性听力障碍是中国人群中主要且常见的出生缺陷之一。非综合征性感音神经性听力损失(NSHL)是遗传性听力障碍最常见的类型。在基因型和表型上,NSHL具有高度异质性,遵循常染色体显性、常染色体隐性或X连锁遗传模式。目前,已有127个基因被确定与综合征性和非综合征性听力损失均相关。在此,我们研究了一个有中度和重度听力障碍的中国家庭。先证者是一名30岁的中国男性。先证者出生时听力正常,5岁时首次被发现有听力障碍。到30岁时,先证者双耳听力逐渐且持续丧失。先证者与其弟弟在临床症状、听力丧失的发病年龄或进展方面相似。先证者的父母表型正常且非近亲结婚。先证者及其弟弟的临床诊断通过经典纯音听力图(PTA)完成。计算机断层扫描(CT)显示双侧外耳、中耳和内耳均无异常。使用一组据报道与遗传性听力障碍相关的127个基因进行靶向二代测序。在先证者及其弟弟中发现了该基因第4外显子中的一个新的纯合单核苷酸缺失(c.427delT)。桑格测序证实先证者的父亲和母亲以杂合方式携带此突变。在100名正常健康对照个体中未发现此突变。该突变(c.427delT)导致移码(p.Tyr143Ilefs19),从而形成一个162个氨基酸的截短的ILDR1蛋白,而不是野生型546个氨基酸的ILDR1蛋白。相关的遗传性听力障碍非常罕见,这是在中国人群中首次报道在与遗传性听力障碍相关的基因中鉴定出突变。我们目前的研究还强调了通过靶向二代测序对遗传性听力障碍患者进行快速且准确、经济有效的筛查的重要性。