Bravin Carlo, Licini Giulia, Hunter Christopher A, Zonta Cristiano
Department of Chemical Sciences , University of Padova , Via Marzolo 1 , 35131 Padova , Italy . Email:
Department of Chemistry , University of Cambridge , Lensfield Road , Cambridge CB2 1EW , UK.
Chem Sci. 2018 Nov 22;10(5):1466-1471. doi: 10.1039/c8sc04406f. eCollection 2019 Feb 7.
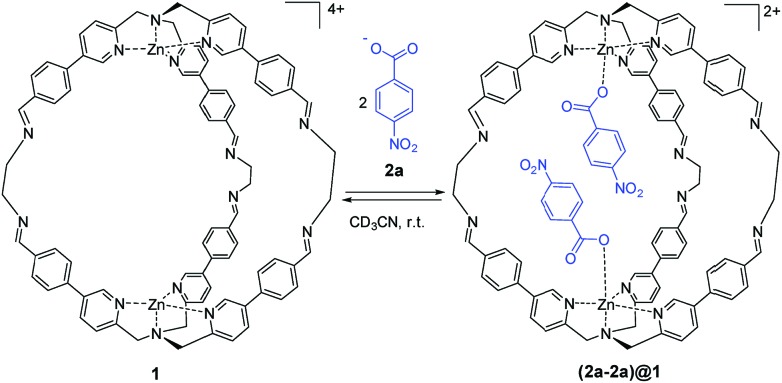
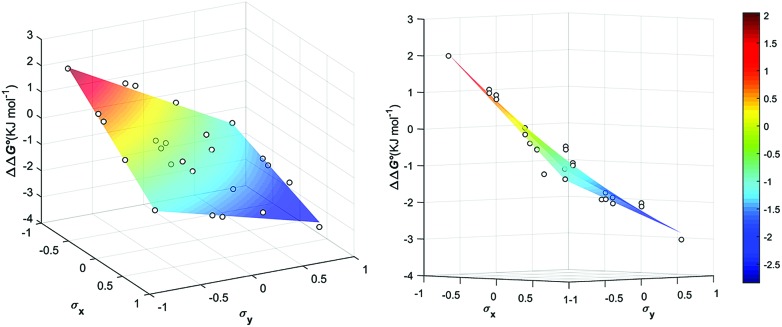
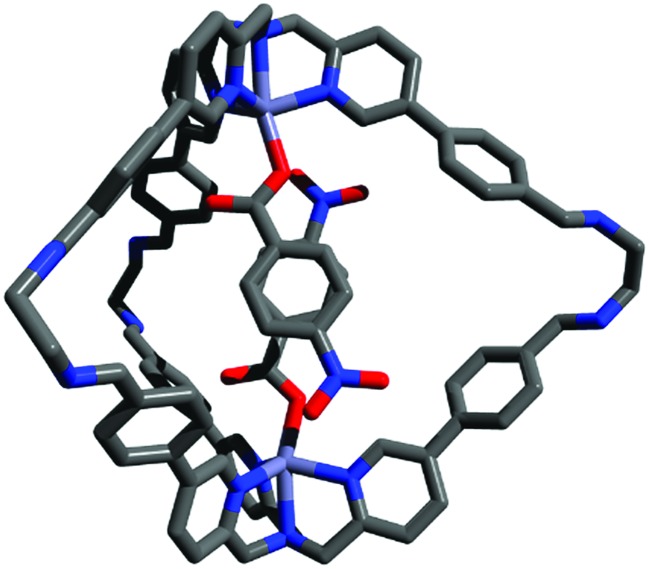
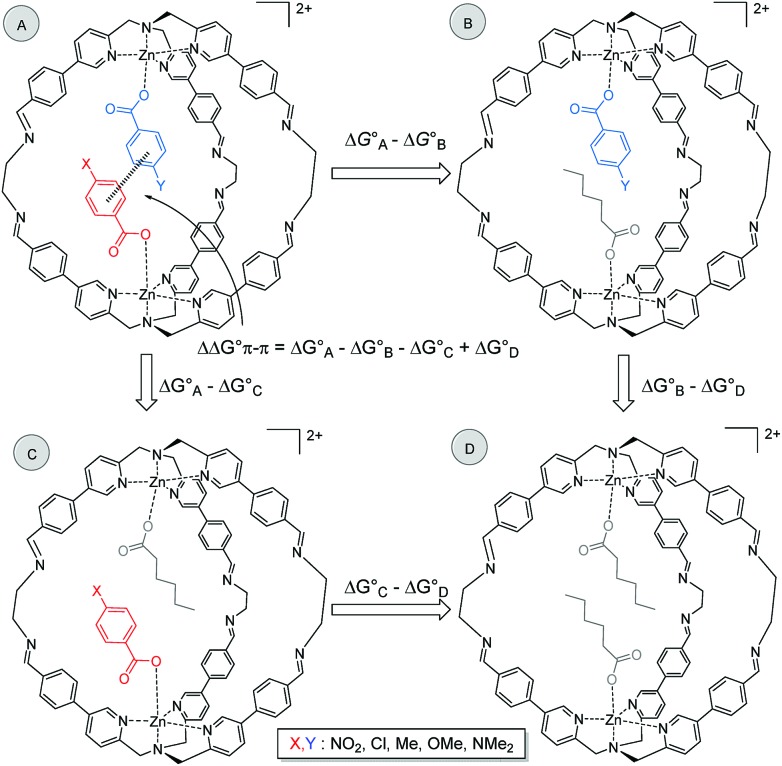
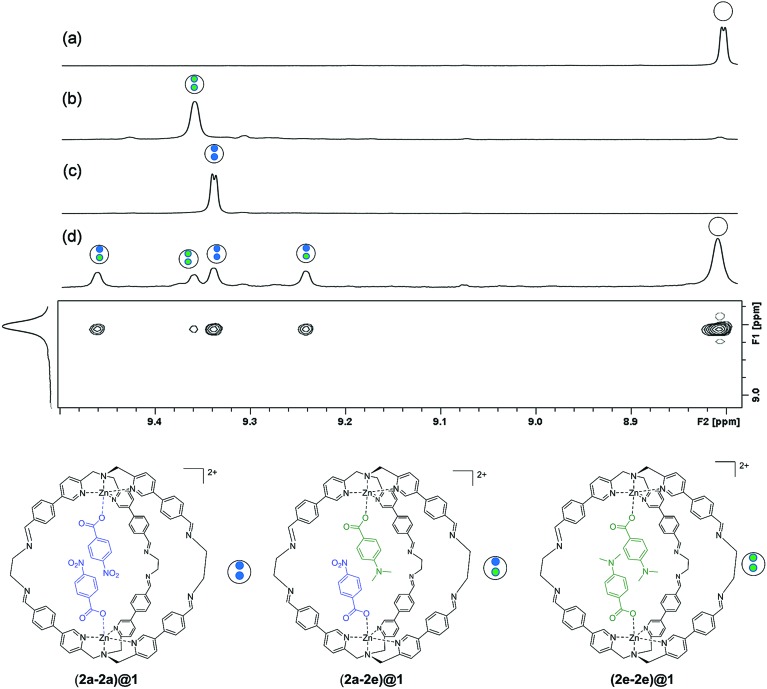
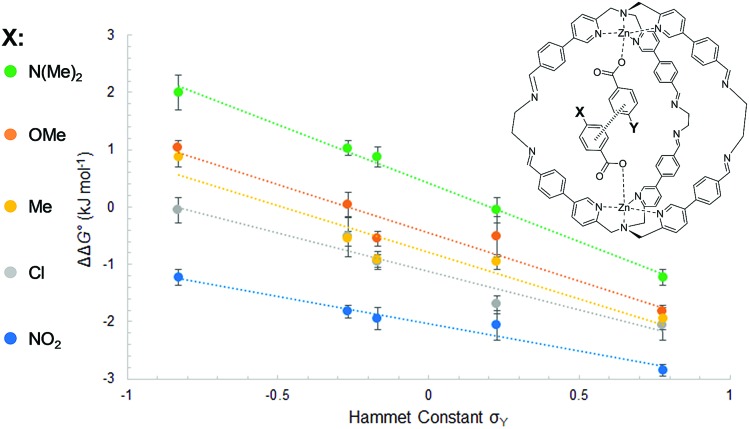
The widespread presence of aromatic stacking interactions in chemical and biological systems, combined with their relatively small energetic contribution, have led to a plethora of theoretical and experimental studies for their quantification and rationalization. Typically, π-π aromatic interactions are studied as a function of substituents to gather information about the interaction mechanism. While experiments suggest that aromatic interactions are dominated by local electrostatic contacts between π-electron density and CH groups, theoretical work has raised the possibility that direct electrostatic interactions between local dipoles of the substituents may play a role. We describe a supramolecular cage that binds two aromatic carboxylates in a stacked geometry such that the aromatic substituents are remote in space. Chemical Double Mutant Cycles (DMCs) were used to measure fifteen different aromatic stacking interactions as a function of substituent (NMe, OMe, Me, Cl and NO). When both aromatic rings have electron-withdrawing nitro substituents, the interaction is attractive (-2.8 kJ mol) due to reduced π-electron repulsion. When both aromatic rings have electron-donating di-methylamino substituents, the interaction is repulsive (+2.0 kJ mol) due to increased π-electron repulsion. The results show that aromatic stacking interactions are dominated by short range electrostatic contacts rather than substituent dipole interactions.
芳香堆积相互作用在化学和生物系统中广泛存在,加之其能量贡献相对较小,引发了大量关于其定量和合理化的理论与实验研究。通常,π-π芳香相互作用作为取代基的函数进行研究,以获取有关相互作用机制的信息。虽然实验表明芳香相互作用主要由π电子密度与CH基团之间的局部静电接触主导,但理论研究提出取代基局部偶极之间的直接静电相互作用可能发挥作用的可能性。我们描述了一种超分子笼,它以堆叠几何结构结合两个芳香羧酸盐,使得芳香取代基在空间上相距较远。化学双突变循环(DMCs)用于测量十五种不同的芳香堆积相互作用作为取代基(NMe、OMe、Me、Cl和NO)的函数。当两个芳香环都具有吸电子硝基取代基时,由于π电子排斥减少,相互作用是吸引性的(-2.8 kJ/mol)。当两个芳香环都具有供电子二甲基氨基取代基时,由于π电子排斥增加,相互作用是排斥性的(+2.0 kJ/mol)。结果表明,芳香堆积相互作用主要由短程静电接触而非取代基偶极相互作用主导。