China University of Mining and Technology, Xuzhou 221116, China.
College of Information Science and Engineering, Zaozhuang University, Zaozhuang 277100, Shandong, China.
Int J Mol Sci. 2019 Feb 23;20(4):978. doi: 10.3390/ijms20040978.
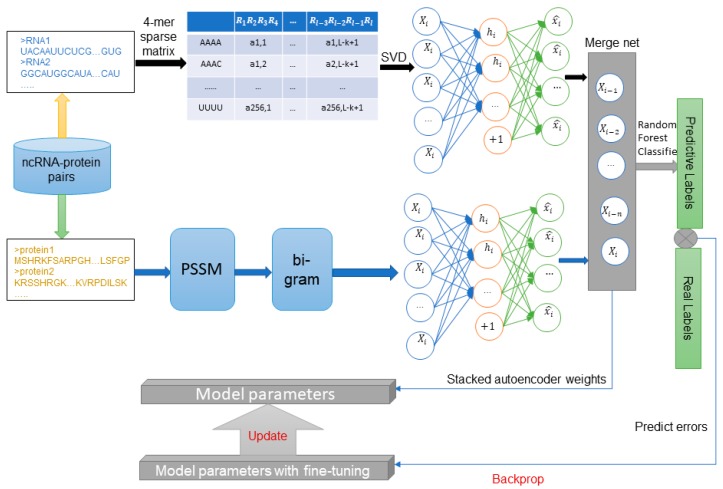
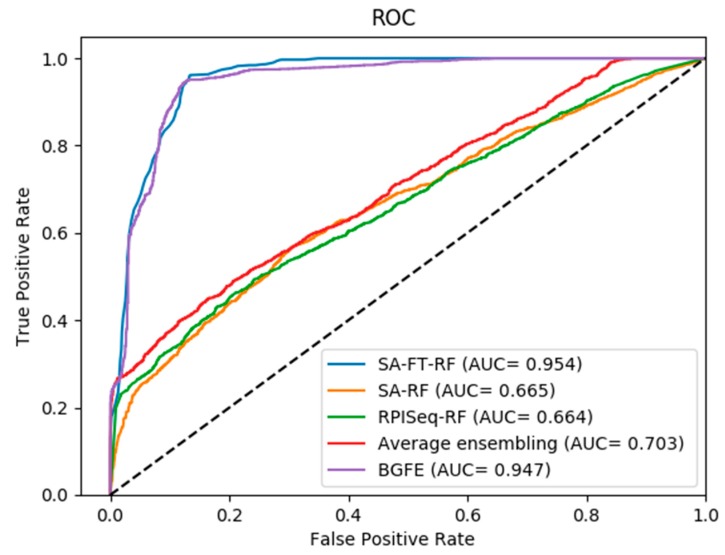
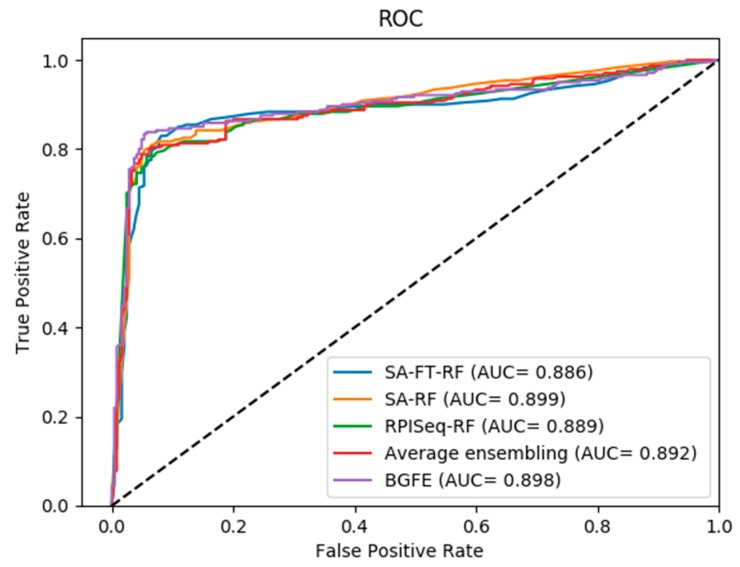
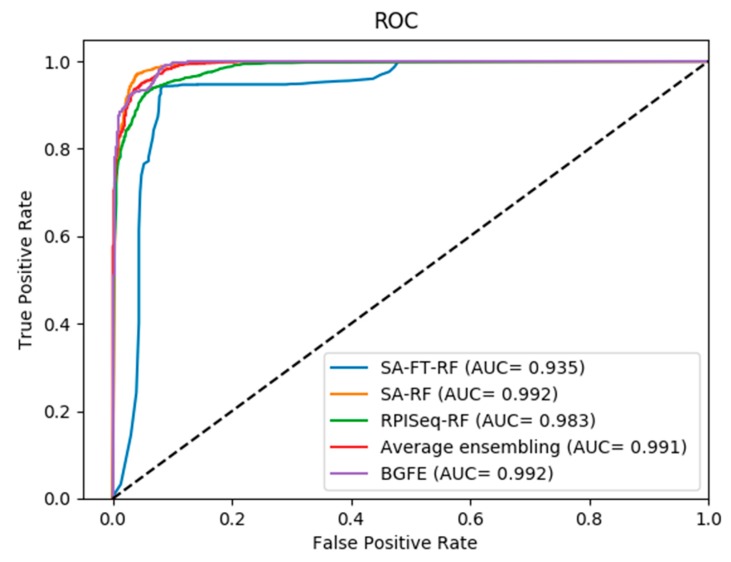
The interactions between ncRNAs and proteins are critical for regulating various cellular processes in organisms, such as gene expression regulations. However, due to limitations, including financial and material consumptions in recent experimental methods for predicting ncRNA and protein interactions, it is essential to propose an innovative and practical approach with convincing performance of prediction accuracy. In this study, based on the protein sequences from a biological perspective, we put forward an effective deep learning method, named BGFE, to predict ncRNA and protein interactions. Protein sequences are represented by bi-gram probability feature extraction method from Position Specific Scoring Matrix (PSSM), and for ncRNA sequences, k-mers sparse matrices are employed to represent them. Furthermore, to extract hidden high-level feature information, a stacked auto-encoder network is employed with the stacked ensemble integration strategy. We evaluate the performance of the proposed method by using three datasets and a five-fold cross-validation after classifying the features through the random forest classifier. The experimental results clearly demonstrate the effectiveness and the prediction accuracy of our approach. In general, the proposed method is helpful for ncRNA and protein interacting predictions and it provides some serviceable guidance in future biological research.
ncRNA 和蛋白质之间的相互作用对于调节生物体的各种细胞过程至关重要,例如基因表达调控。然而,由于最近预测 ncRNA 和蛋白质相互作用的实验方法在财务和材料消耗方面存在限制,因此提出一种具有创新性和实用性的方法,具有令人信服的预测准确性表现至关重要。在这项研究中,我们从生物角度出发,基于蛋白质序列,提出了一种有效的深度学习方法,称为 BGFE,用于预测 ncRNA 和蛋白质相互作用。蛋白质序列通过位置特异性评分矩阵 (PSSM) 的双元概率特征提取方法表示,而对于 ncRNA 序列,则使用 k-mer 稀疏矩阵来表示。此外,为了提取隐藏的高级特征信息,我们使用堆叠自动编码器网络和堆叠集成策略。通过随机森林分类器对特征进行分类后,我们使用三个数据集和五折交叉验证来评估所提出方法的性能。实验结果清楚地表明了我们方法的有效性和预测准确性。总的来说,该方法有助于 ncRNA 和蛋白质相互作用的预测,并为未来的生物研究提供了一些有用的指导。