Department of Chemistry and Molecular Biology, University of Gothenburg, 405 30, Göteborg, Sweden.
Apoptosis Research Centre, National University of Ireland Galway, Galway, Ireland.
Sci Rep. 2019 Mar 4;9(1):3407. doi: 10.1038/s41598-019-39939-z.
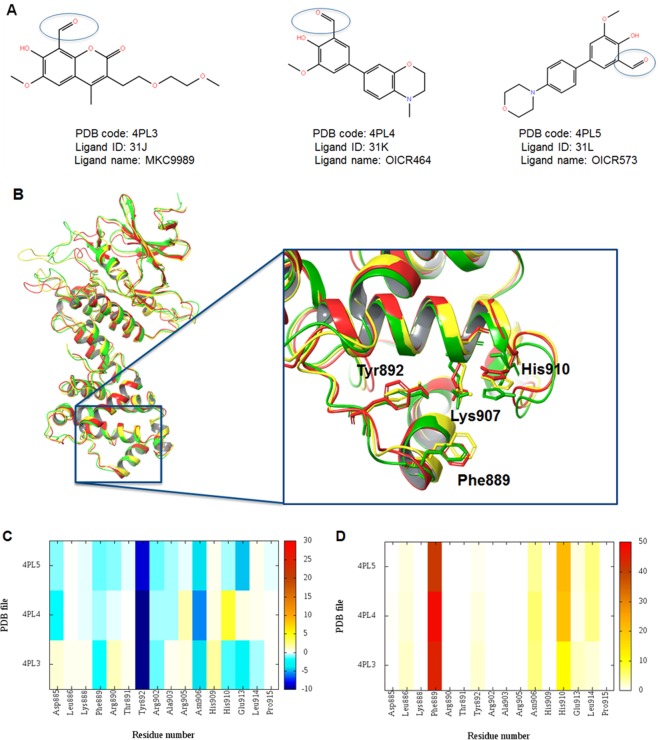
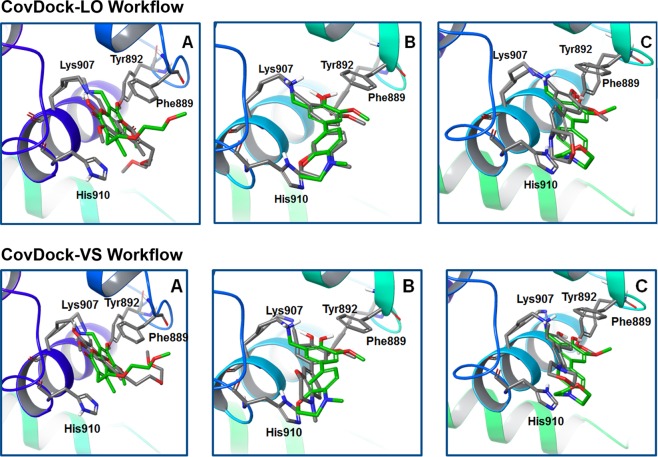
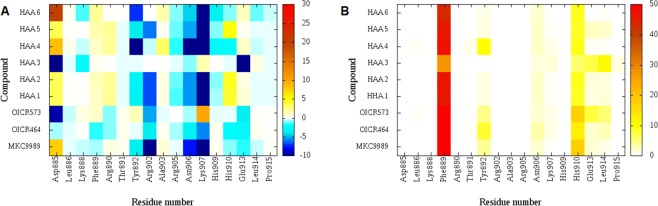
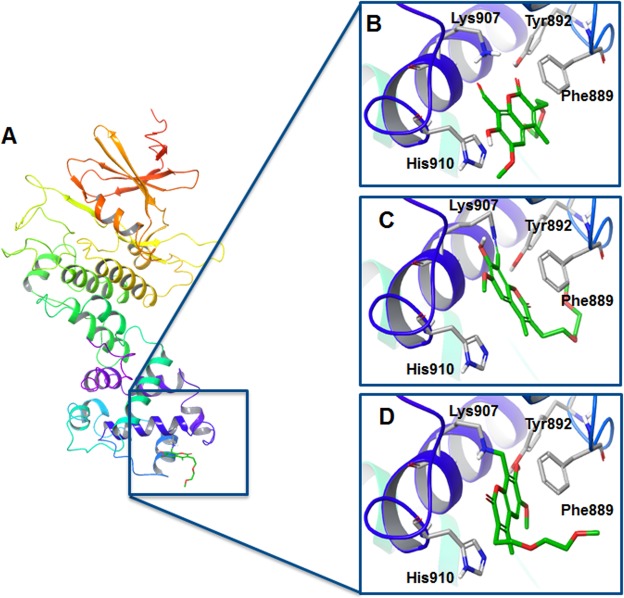
IRE1 is an endoplasmic reticulum (ER) bound transmembrane bifunctional kinase and endoribonuclease protein crucial for the unfolded protein response (UPR) signaling pathway. Upon ER stress, IRE1 homodimerizes, oligomerizes and autophosphorylates resulting in endoribonuclease activity responsible for excision of a 26 nucleotide intron from the X-box binding protein 1 (XBP1) mRNA. This unique splicing mechanism results in activation of the XBP1s transcription factor to specifically restore ER stress. Small molecules targeting the reactive lysine residue (Lys907) in IRE1α's RNase domain have been shown to inhibit the cleavage of XBP1 mRNA. Crystal structures of murine IRE1 in complex with covalently bound hydroxyl aryl aldehyde (HAA) inhibitors show that these molecules form hydrophobic interactions with His910 and Phe889, a hydrogen bond with Tyr892 and an indispensable Schiff-base with Lys907. The availability of such data prompted interest in exploring structure-based drug design as a strategy to develop new covalently binding ligands. We extensively evaluated conventional and covalent docking for drug discovery targeting the catalytic site of the RNase domain. The results indicate that neither computational approach is fully successful in the current case, and we highlight herein the potential and limitations of the methods for the design of novel IRE1 RNase binders.
IRE1 是内质网 (ER) 结合的跨膜双功能激酶和内切核糖核酸酶蛋白,对未折叠蛋白反应 (UPR) 信号通路至关重要。在 ER 应激时,IRE1 同二聚化、寡聚化并自磷酸化,导致内切核糖核酸酶活性负责从 X 盒结合蛋白 1 (XBP1) mRNA 中切除 26 个核苷酸的内含子。这种独特的剪接机制导致 XBP1s 转录因子的激活,以特异性恢复 ER 应激。靶向 IRE1α 的 RNase 结构域中反应性赖氨酸残基 (Lys907) 的小分子已被证明可以抑制 XBP1 mRNA 的切割。与共价结合的羟基芳基醛 (HAA) 抑制剂的鼠 IRE1 的晶体结构表明,这些分子与 His910 和 Phe889 形成疏水性相互作用,与 Tyr892 形成氢键,并与 Lys907 形成不可缺少的席夫碱。有了这些数据,人们就有兴趣探索基于结构的药物设计作为开发新的共价结合配体的策略。我们广泛评估了针对 RNase 结构域催化位点的药物发现的常规和共价对接。结果表明,这两种计算方法在目前的情况下都不完全成功,我们在此强调了这些方法在设计新型 IRE1 RNase 结合物方面的潜力和局限性。