Chen Qun, Thompson Jeremy, Hu Ying, Das Anindita, Lesnefsky Edward J
Division of Cardiology, Departments of Medicine, Virginia Commonwealth University, Richmond, VA, United States.
Biochemistry and Molecular Biology, Virginia Commonwealth University, Richmond, VA, United States.
Front Cardiovasc Med. 2019 Feb 19;6:10. doi: 10.3389/fcvm.2019.00010. eCollection 2019.
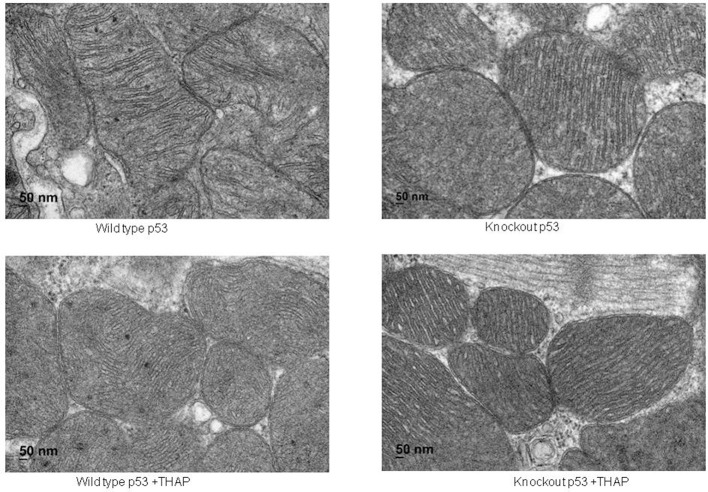
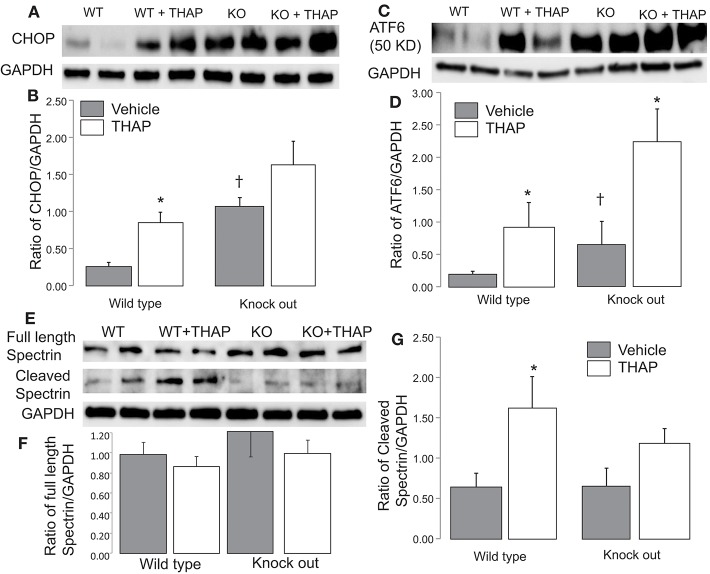
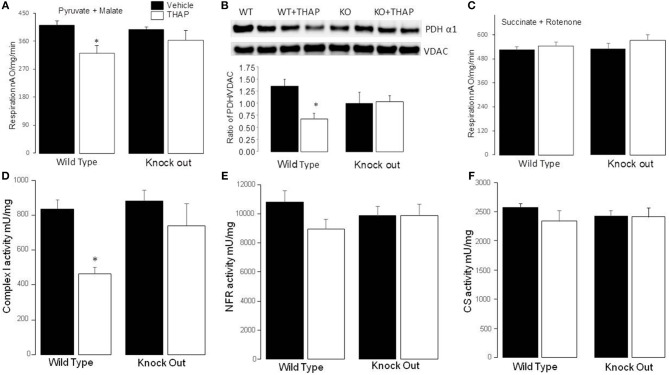
Endoplasmic reticulum (ER) stress contributes to cardiovascular disease including heart failure. Interactions between the ER and mitochondria during ER stress can impair the mitochondrial respiratory chain and increase cell injury. p53 is a tumor suppressor protein that regulates apoptosis. p53 contributes to the regulation of mitochondrial and ER interactions, especially during the progression of ER stress. The knockout (KO) of p53 leads to decreased injury in hearts following ischemia-reperfusion. We asked if KO of p53 can protect mitochondria during the induction of ER stress and decrease cell injury. Floxed p53 mice were crossed with mice carrying an α-myosin heavy chain to generate cardiac specific p53 KO mice. Thapsigargin (THAP) was used to induce ER stress in wild type (WT) and p53 KO mice. Mice were euthanized after 48 h THAP treatment. Cardiac mitochondria were isolated for functional measurement. TUNEL staining was used to assess myocyte death. In WT mice, THAP treatment decreased the rate of oxidative phosphorylation using pyruvate + malate as complex I substrates compared to vehicle-treated control. Complex I activity was also decreased in the THAP-treated WT mice. The rate of oxidative phosphorylation and complex I activity were not altered in THAP-treated p53 KO mice. The content of pyruvate dehydrogenase (PDH) α1 subunit was decreased in THAP-treated WT mice but not in p53 KO mice. ER stress led to a release of cytochrome and apoptosis inducing factor from mitochondria into cytosol in WT but not in KO mice. Knockout of p53 also preserved mitochondrial bcl-2 content in THAP-treated mice. In WT mice, THAP treatment markedly increased cell death compared to vehicle treated hearts. In contrast, cell injury was decreased in THAP-treated p53 KO mice compared to corresponding wild type. Thus, KO of p53 decreased cell injury by protecting mitochondria during the ER stress.
内质网(ER)应激与包括心力衰竭在内的心血管疾病有关。内质网应激期间内质网与线粒体之间的相互作用会损害线粒体呼吸链并增加细胞损伤。p53是一种调节细胞凋亡的肿瘤抑制蛋白。p53有助于调节线粒体与内质网的相互作用,尤其是在内质网应激进展过程中。p53基因敲除(KO)可减少缺血再灌注后心脏的损伤。我们研究了p53基因敲除是否能在内质网应激诱导过程中保护线粒体并减少细胞损伤。将携带floxed p53的小鼠与携带α-肌球蛋白重链的小鼠杂交,以产生心脏特异性p53基因敲除小鼠。用毒胡萝卜素(THAP)诱导野生型(WT)和p53基因敲除小鼠的内质网应激。THAP处理48小时后对小鼠实施安乐死。分离心脏线粒体进行功能测定。采用TUNEL染色评估心肌细胞死亡情况。在野生型小鼠中,与溶媒处理的对照组相比,THAP处理降低了以丙酮酸+苹果酸作为复合体I底物时的氧化磷酸化速率。THAP处理的野生型小鼠中复合体I活性也降低。THAP处理的p53基因敲除小鼠的氧化磷酸化速率和复合体I活性未改变。THAP处理的野生型小鼠中丙酮酸脱氢酶(PDH)α1亚基的含量降低,但p53基因敲除小鼠中未降低。内质网应激导致野生型小鼠而非基因敲除小鼠的线粒体中细胞色素c和凋亡诱导因子释放到细胞质中。p53基因敲除还能维持THAP处理小鼠的线粒体bcl-2含量。在野生型小鼠中,与溶媒处理的心脏相比,THAP处理显著增加了细胞死亡。相比之下,与相应的野生型相比,THAP处理的p53基因敲除小鼠的细胞损伤减少。因此,p53基因敲除通过在内质网应激期间保护线粒体来减少细胞损伤。