Center for Neurodegenerative Science, Van Andel Research Institute, Grand Rapids, MI 49503.
Department of Translational Science and Molecular Medicine, Michigan State University, Grand Rapids, MI 49503.
Proc Natl Acad Sci U S A. 2019 Mar 19;116(12):5765-5774. doi: 10.1073/pnas.1814909116. Epub 2019 Mar 6.
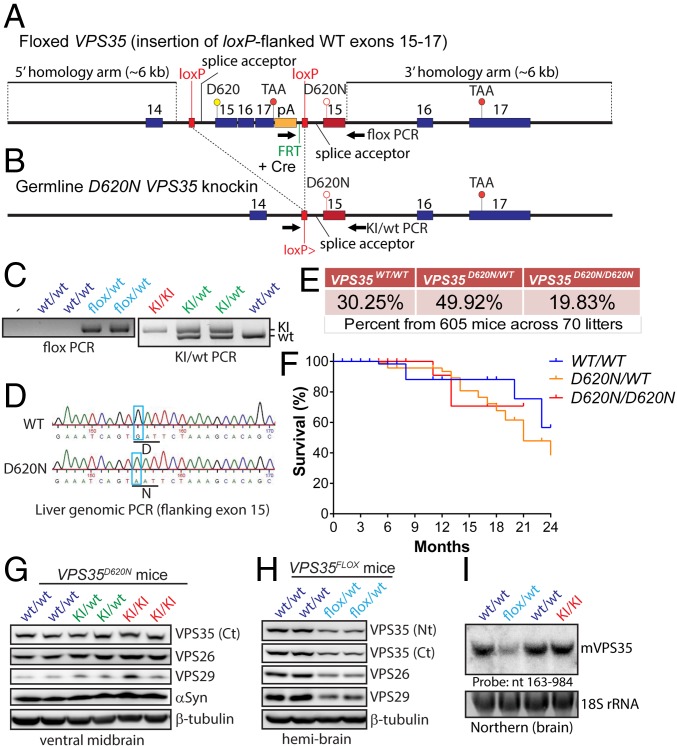
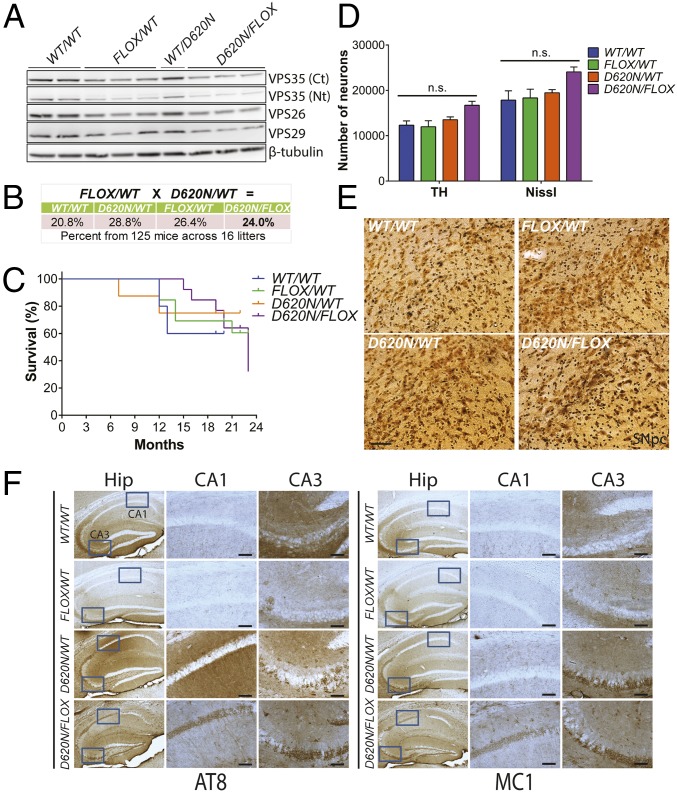
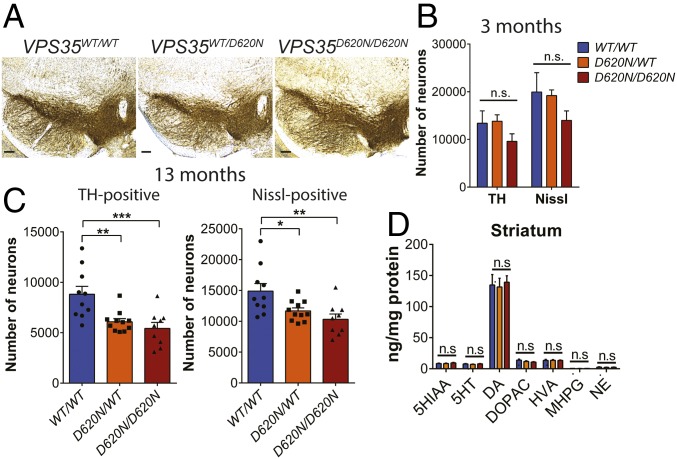
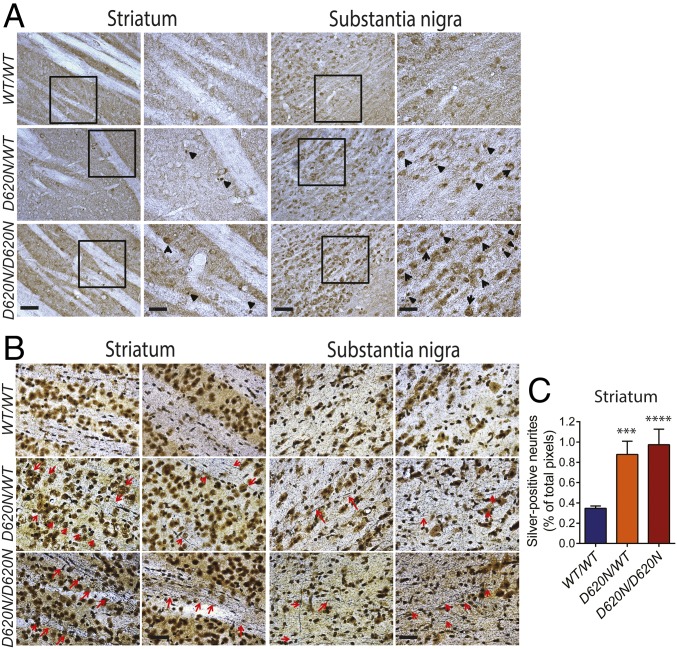
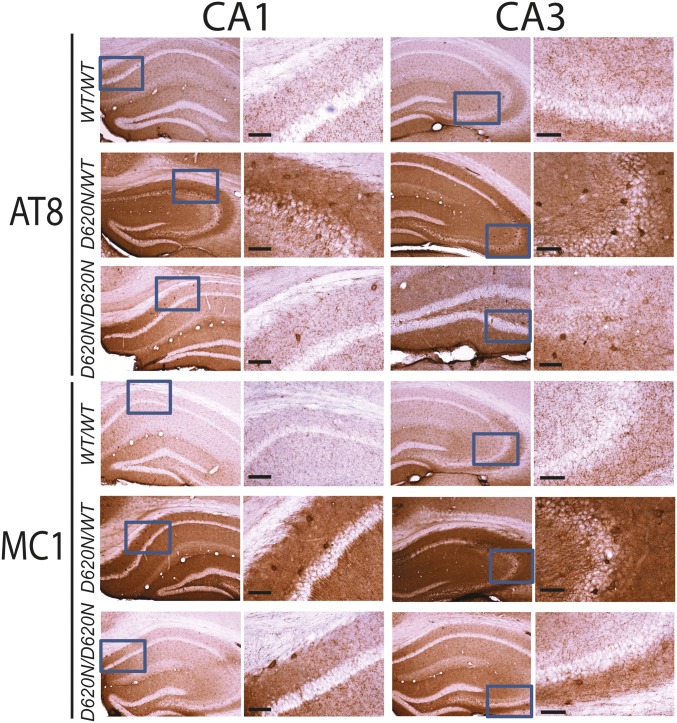
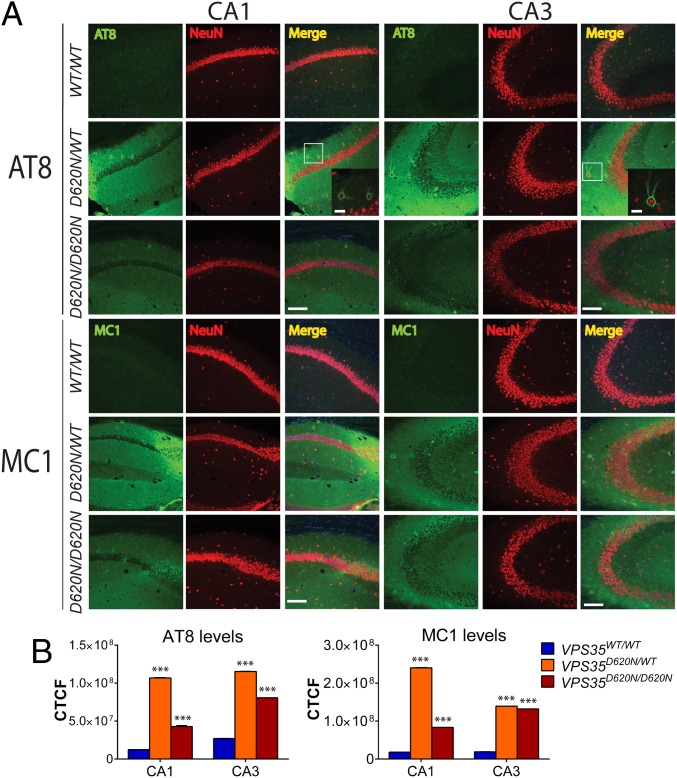
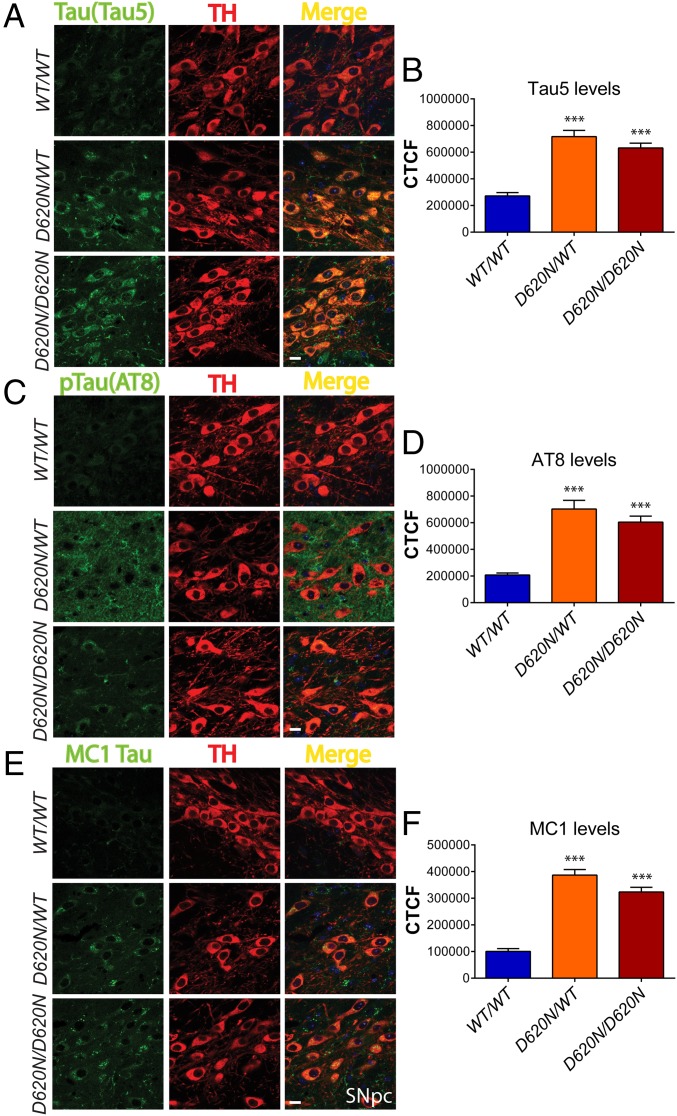
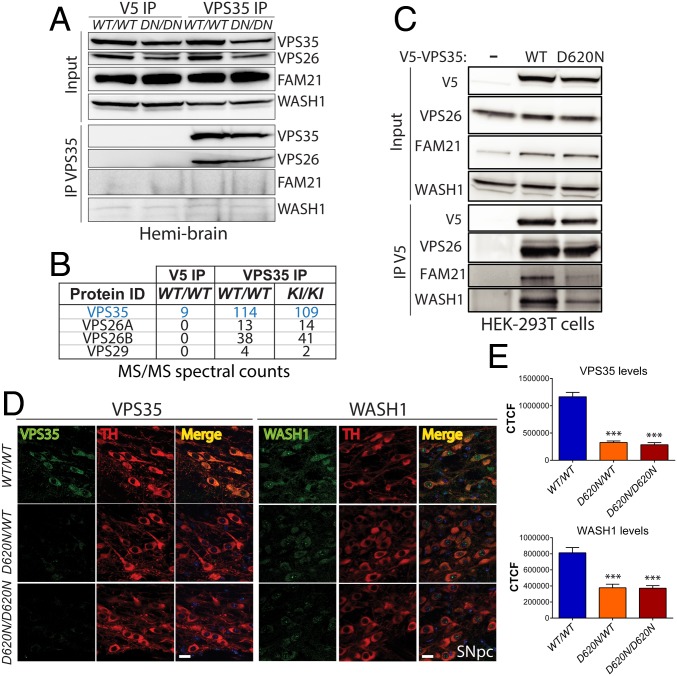
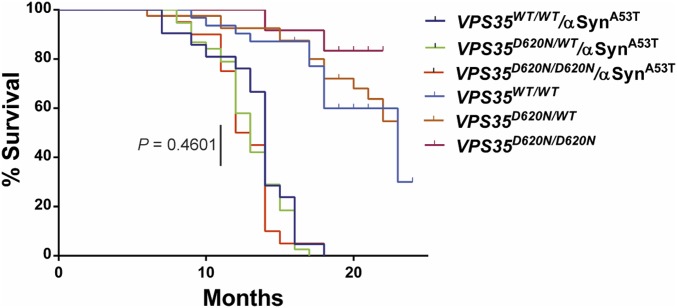
Mutations in the () gene represent a cause of late-onset, autosomal dominant familial Parkinson's disease (PD). A single missense mutation, D620N, is considered pathogenic based upon its segregation with disease in multiple families with PD. At present, the mechanism(s) by which familial mutations precipitate neurodegeneration in PD are poorly understood. Here, we employ a germline knockin (KI) mouse model of PD to formally establish the age-related pathogenic effects of the D620N mutation at physiological expression levels. Our data demonstrate that a heterozygous or homozygous D620N mutation is sufficient to reproduce key neuropathological hallmarks of PD as indicated by the progressive degeneration of nigrostriatal pathway dopaminergic neurons and widespread axonal pathology. Unexpectedly, endogenous D620N VPS35 expression induces robust tau-positive somatodendritic pathology throughout the brain as indicated by abnormal hyperphosphorylated and conformation-specific tau, which may represent an important and early feature of mutant VPS35-induced neurodegeneration in PD. In contrast, we find no evidence for α-synuclein-positive neuropathology in aged KI mice, a hallmark of Lewy body pathology in PD. D620N VPS35 expression also fails to modify the lethal neurodegenerative phenotype of human A53T-α-synuclein transgenic mice. Finally, by crossing KI and null mice, our data demonstrate that a single allele is sufficient for survival and early maintenance of dopaminergic neurons, indicating that the D620N VPS35 protein is fully functional. Our data raise the tantalizing possibility of a pathogenic interplay between mutant VPS35 and tau for inducing neurodegeneration in PD.
()基因突变是导致迟发性、常染色体显性家族性帕金森病(PD)的一个原因。单一错义突变 D620N 被认为是致病的,因为它在多个 PD 家族中与疾病共分离。目前,家族性突变如何导致 PD 中的神经退行性变的机制尚不清楚。在这里,我们使用 PD 的种系敲入(KI)小鼠模型来正式建立 D620N 突变在生理表达水平下与年龄相关的致病效应。我们的数据表明,杂合或纯合 D620N 突变足以复制 PD 的关键神经病理学特征,如黑质纹状体通路多巴胺能神经元的进行性退化和广泛的轴突病理学。出乎意料的是,内源性 D620N VPS35 表达会诱导整个大脑中强壮的 tau 阳性体树突病理学,如异常高磷酸化和构象特异性 tau 所表明的,这可能代表突变 VPS35 诱导 PD 神经退行性变的一个重要和早期特征。相比之下,我们在老年 KI 小鼠中没有发现α-突触核蛋白阳性神经病理学的证据,这是 PD 路易体病理学的一个标志。D620N VPS35 表达也不能改变人类 A53T-α-突触核蛋白转基因小鼠的致命神经退行性表型。最后,通过交叉 KI 和 null 小鼠,我们的数据表明,单个 等位基因足以维持多巴胺能神经元的存活和早期维持,表明 D620N VPS35 蛋白是完全功能的。我们的数据提出了一个诱人的可能性,即突变 VPS35 和 tau 之间存在致病相互作用,可能导致 PD 中的神经退行性变。