Department of Neurodegenerative Science, Van Andel Institute, 333 Bostwick Ave NE, Grand Rapids, MI, 49503, USA.
Brain Mind Institute, School of Life Sciences, Swiss Federal Institute of Technology (EPFL), Lausanne, Vaud, 1015, Switzerland.
Mol Neurodegener. 2023 Aug 4;18(1):51. doi: 10.1186/s13024-023-00641-4.
Mutations in the vacuolar protein sorting 35 ortholog (VPS35) gene cause late-onset, autosomal dominant Parkinson's disease (PD), with a single missense mutation (Asp620Asn, D620N) known to segregate with disease in families with PD. The VPS35 gene encodes a core component of the retromer complex, involved in the endosomal sorting and recycling of transmembrane cargo proteins. VPS35-linked PD is clinically indistinguishable from sporadic PD, although it is not yet known whether VPS35-PD brains exhibit α-synuclein-positive brainstem Lewy pathology that is characteristic of sporadic cases. Prior studies have suggested a functional interaction between VPS35 and the PD-linked gene product α-synuclein in lower organisms, where VPS35 deletion enhances α-synuclein-induced toxicity. In mice, VPS35 overexpression is reported to rescue hippocampal neuronal loss in human α-synuclein transgenic mice, potentially suggesting a retromer deficiency in these mice.
Here, we employ multiple well-established genetic rodent models to explore a functional or pathological interaction between VPS35 and α-synuclein in vivo.
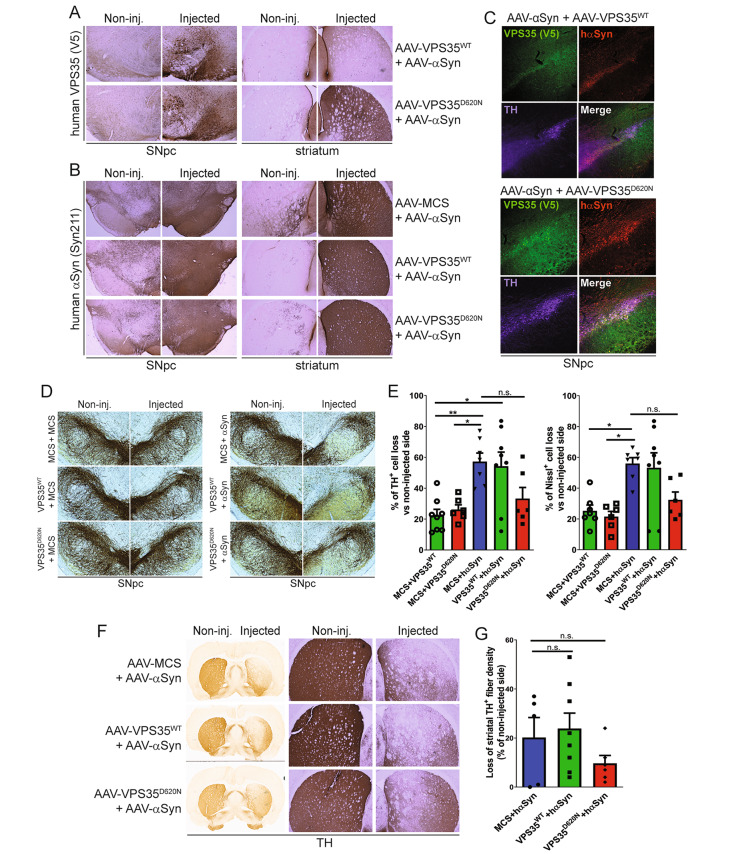
We find that endogenous α-synuclein is dispensable for nigrostriatal pathway dopaminergic neurodegeneration induced by the viral-mediated delivery of human D620N VPS35 in mice, suggesting that α-synuclein does not operate downstream of VPS35. We next evaluated retromer levels in affected brain regions from human A53T-α-synuclein transgenic mice, but find normal levels of the core subunits VPS35, VPS26 or VPS29. We further find that heterozygous VPS35 deletion fails to alter the lethal neurodegenerative phenotype of these A53T-α-synuclein transgenic mice, suggesting the absence of retromer deficiency in this PD model. Finally, we explored the neuroprotective capacity of increasing VPS35 expression in a viral-based human wild-type α-synuclein rat model of PD. However, we find that the overexpression of wild-type VPS35 is not sufficient for protection against α-synuclein-induced nigral dopaminergic neurodegeneration, α-synuclein pathology and reactive gliosis.
Collectively, our data suggest a limited interaction of VPS35 and α-synuclein in neurodegenerative models of PD, and do not provide support for their interaction within a common pathophysiological pathway.
液泡蛋白分选 35 同源物(VPS35)基因突变导致晚发性常染色体显性帕金森病(PD),已知家族性 PD 中存在单一错义突变(Asp620Asn,D620N)与疾病分离。VPS35 基因编码参与跨膜货物蛋白内体分拣和回收的逆行体复合物的核心组成部分。VPS35 相关 PD 在临床上与散发性 PD 无法区分,尽管目前尚不清楚 VPS35-PD 大脑是否存在特征性散发性病例的α-突触核蛋白阳性脑干路易体病理学。先前的研究表明,VPS35 和 PD 相关基因产物α-突触核蛋白在低等生物中存在功能相互作用,其中 VPS35 缺失增强了α-突触核蛋白诱导的毒性。在小鼠中,据报道 VPS35 过表达可挽救人类α-突触核蛋白转基因小鼠的海马神经元丢失,这可能表明这些小鼠存在逆行体缺陷。
在这里,我们采用多种成熟的遗传啮齿动物模型来探索 VPS35 和α-突触核蛋白在体内的功能或病理相互作用。
我们发现内源性α-突触核蛋白对于病毒介导的人类 D620N VPS35 传递在小鼠中引起的黑质纹状体通路多巴胺能神经退行性变是可有可无的,这表明α-突触核蛋白不在 VPS35 下游起作用。接下来,我们评估了受影响的脑区中的逆行体水平来自人类 A53T-α-突触核蛋白转基因小鼠,但发现核心亚基 VPS35、VPS26 或 VPS29 的水平正常。我们进一步发现杂合 VPS35 缺失不会改变这些 A53T-α-突触核蛋白转基因小鼠的致死性神经退行性表型,这表明在这种 PD 模型中不存在逆行体缺陷。最后,我们在基于病毒的人类野生型α-突触核蛋白大鼠 PD 模型中探索了增加 VPS35 表达的神经保护能力。然而,我们发现野生型 VPS35 的过表达不足以防止α-突触核蛋白诱导的黑质多巴胺能神经退行性变、α-突触核蛋白病理学和反应性神经胶质增生。
总之,我们的数据表明 VPS35 和α-突触核蛋白在 PD 的神经退行性模型中相互作用有限,并且不支持它们在共同的病理生理途径中的相互作用。