1 Department of Physiology & Pathophysiology School of Basic Medical Sciences Capital Medical University Beijing China.
3 National Clinical Research Center for Geriatric Disorders Xuanwu Hospital of Capital Medical University Beijing China.
J Am Heart Assoc. 2019 Mar 19;8(6):e011179. doi: 10.1161/JAHA.118.011179.
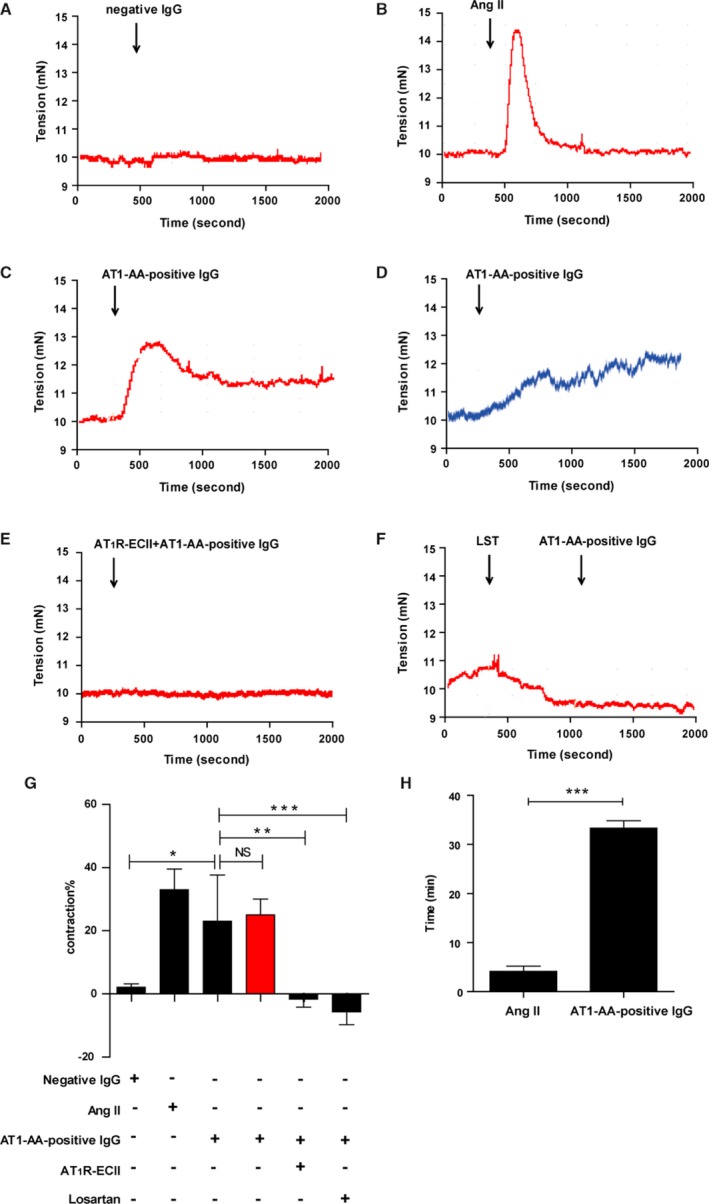
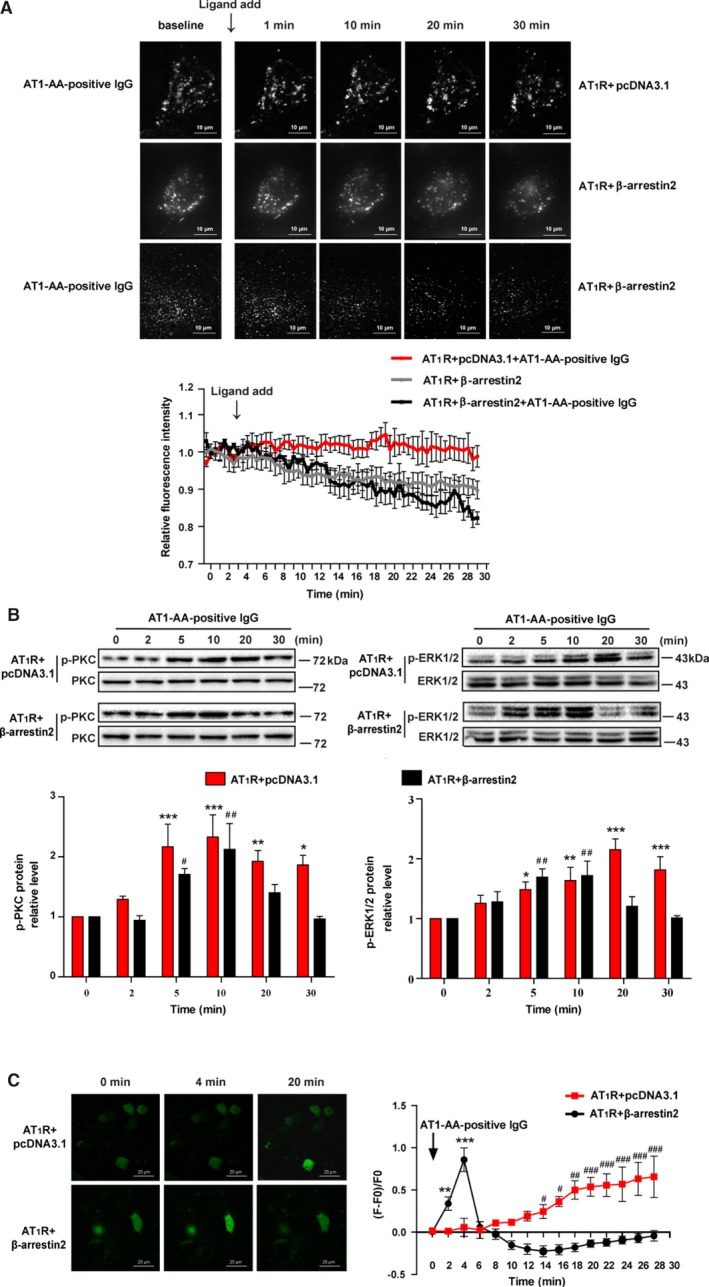
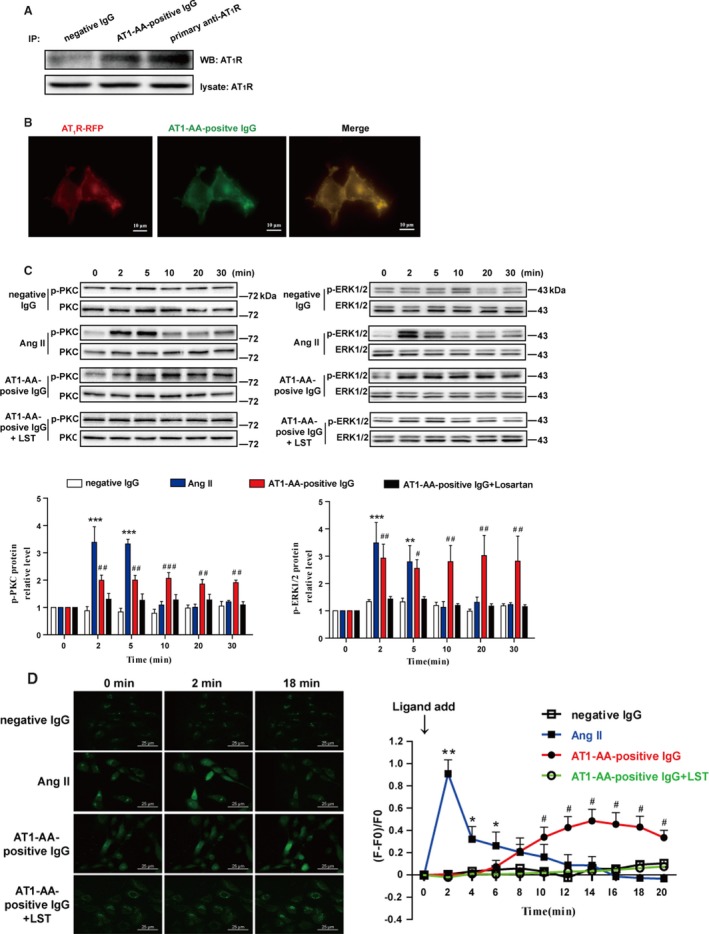
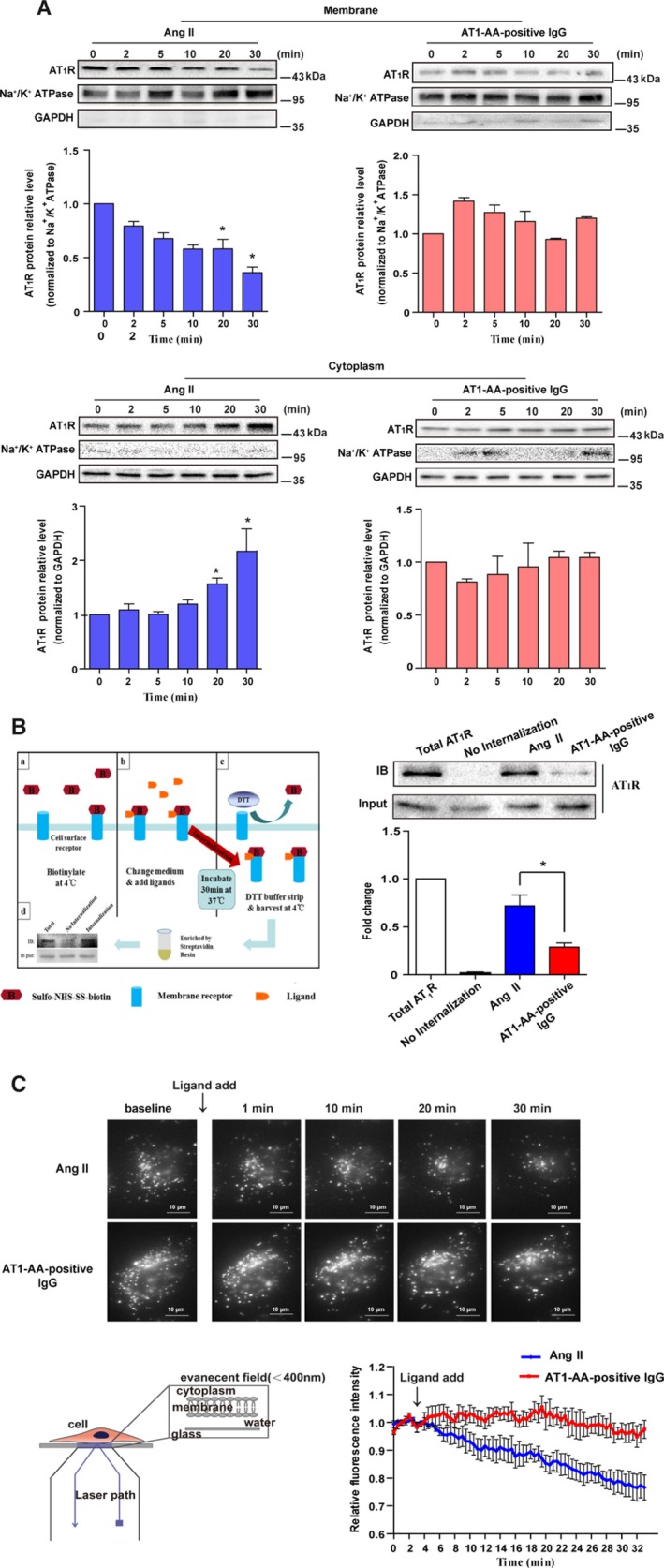
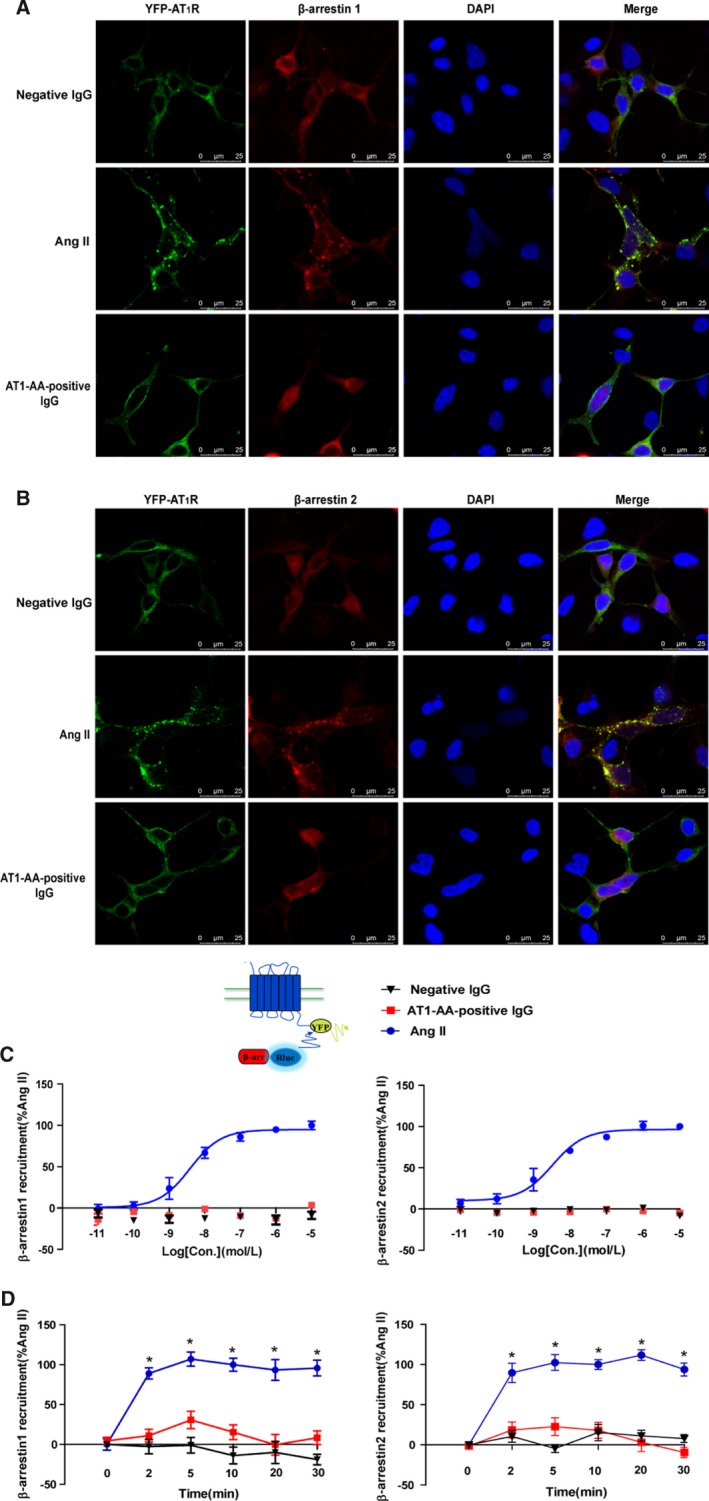
Background Angiotensin II type 1 receptor ( AT R) autoantibody ( AT 1- AA ) was first identified as a causative factor in preeclampsia. Unlike physiological ligand angiotensin II (Ang II ), AT 1- AA can induce vasoconstriction in a sustained manner, causing a series of adverse effects, such as vascular injury and poor placental perfusion. However, its underlying mechanisms remain unclear. Here, from the perspective of AT R internalization, the present study investigated the molecular mechanism of sustained vasoconstriction induced by AT R autoantibody. Methods and Results In the current study, we used the vascular-ring technique to determine that AT 1- AA -positive IgG, which was obtained from the sera of preeclamptic patients, induced long-term vasoconstriction in endothelium-intact or endothelium-denuded rat thoracic arteries. The effect was caused by prolonged activation of AT R downstream signals in vascular smooth muscle cells, including Ca, protein kinase C, and extracellular signal-regulated kinase 1 and 2. Then, using subcellular protein fractionation, cell surface protein biotinylation, and total internal reflection fluorescence, we found that AT 1- AA -positive IgG resulted in significantly less AT R internalization than in the Ang II treatment group. Moreover, through use of fluorescent tracing and bioluminescence resonance energy transfer, we found that AT 1- AA -positive IgG cannot induce the recruitment of β-arrestin1/2, which mediated receptor internalization. Then, the effect of sustained AT R activation induced by AT 1- AA -positive IgG was rescued by overexpression of β-arrestin2. Conclusions These data suggested that limited AT R internalization resulting from the inhibition of β-arrestin1/2 recruitment played an important role in sustained vasoconstriction induced by AT 1- AA -positive IgG, which may set the stage for avoiding AT R overactivation in the management of preeclampsia.
血管紧张素 II 型 1 型受体(AT1R)自身抗体(AT1-AA)最初被确定为先兆子痫的致病因素。与生理配体血管紧张素 II(Ang II)不同,AT1-AA 可以持续引起血管收缩,导致一系列不良影响,如血管损伤和胎盘灌注不良。然而,其潜在机制尚不清楚。在这里,我们从 AT1R 内化的角度,研究了 AT1R 自身抗体引起的持续血管收缩的分子机制。
在目前的研究中,我们使用血管环技术确定来自先兆子痫患者血清的 AT1-AA 阳性 IgG 可引起完整内皮或去内皮大鼠胸主动脉的长期血管收缩。该作用是通过血管平滑肌细胞中 AT1R 下游信号的延长激活引起的,包括 Ca2+、蛋白激酶 C 和细胞外信号调节激酶 1 和 2。然后,通过亚细胞蛋白分级分离、细胞表面蛋白生物素化和全内反射荧光,我们发现 AT1-AA 阳性 IgG 导致 AT1R 内化明显少于 Ang II 处理组。此外,通过荧光示踪和生物发光共振能量转移,我们发现 AT1-AA 阳性 IgG 不能诱导β-arrestin1/2 的募集,后者介导受体内化。然后,通过过表达β-arrestin2 挽救了 AT1-AA 阳性 IgG 引起的持续 AT1R 激活作用。
这些数据表明,AT1-AA 阳性 IgG 引起的 AT1R 内化受限,由于抑制了β-arrestin1/2 的募集,在 AT1-AA 阳性 IgG 引起的持续血管收缩中起重要作用,这可能为避免先兆子痫管理中 AT1R 的过度激活奠定了基础。