Ott Tim, Kaufmann Lilian, Granzow Martin, Hinderhofer Katrin, Bartram Claus R, Theiß Susanne, Seitz Angelika, Paramasivam Nagarajan, Schulz Angela, Moog Ute, Blum Martin, Evers Christina M
Institute of Zoology, University of Hohenheim, Stuttgart, Germany.
Institute of Human Genetics, Heidelberg University, Heidelberg, Germany.
Front Physiol. 2019 Feb 25;10:134. doi: 10.3389/fphys.2019.00134. eCollection 2019.
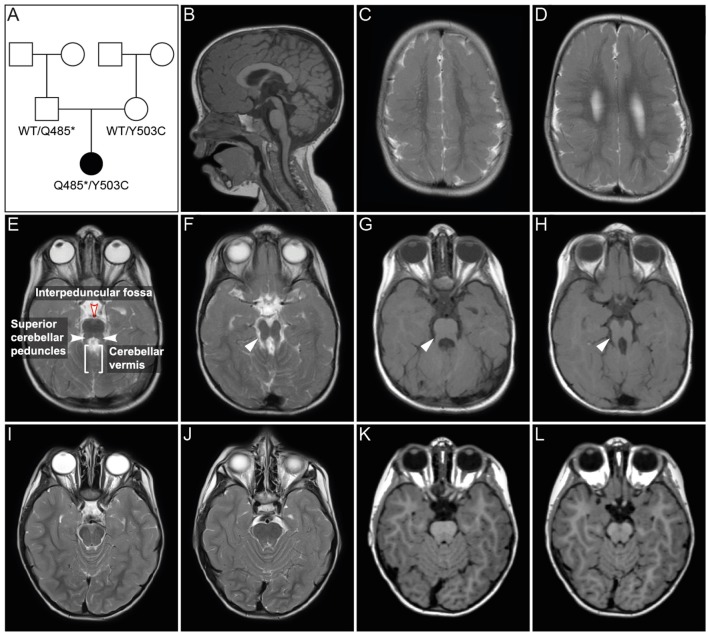
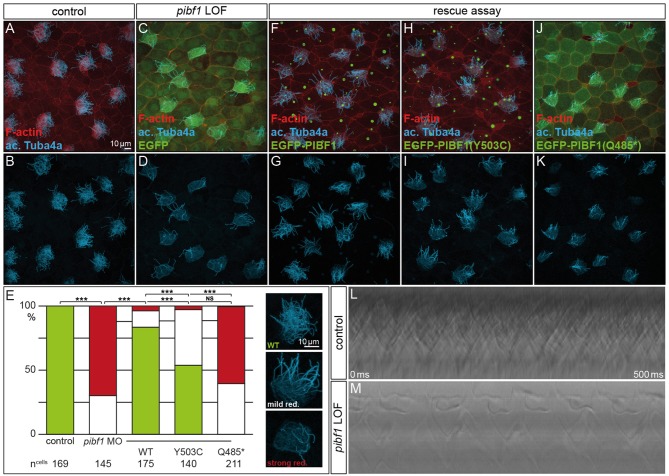
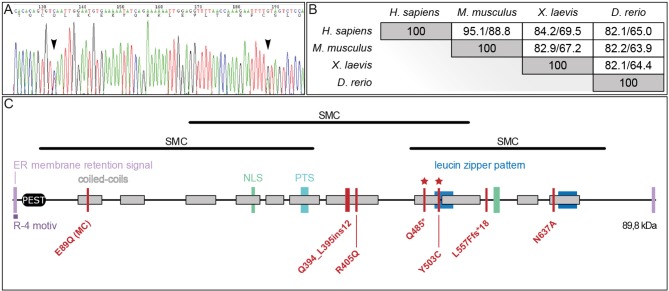
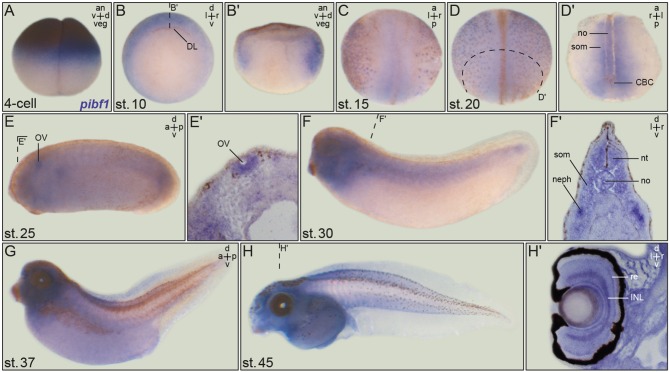
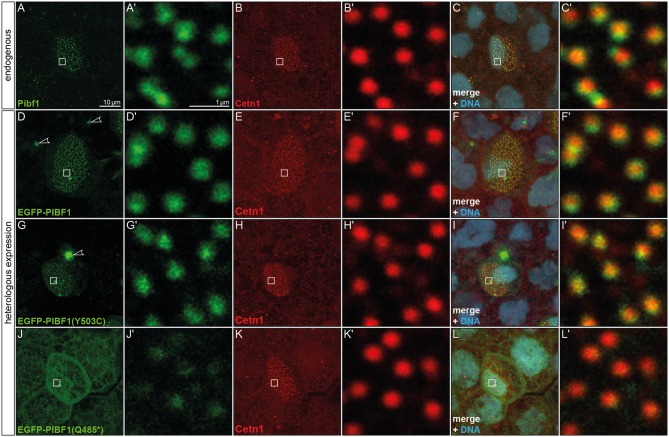
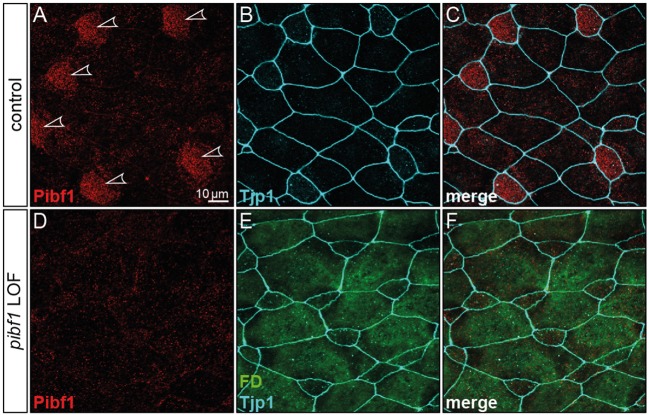
Joubert syndrome (JS) is a congenital autosomal-recessive or-in rare cases-X-linked inherited disease. The diagnostic hallmark of the so-called molar tooth sign describes the morphological manifestation of the mid- and hind-brain in axial brain scans. Affected individuals show delayed development, intellectual disability, ataxia, hyperpnea, sleep apnea, abnormal eye, and tongue movements as well as hypotonia. At the cellular level, JS is associated with the compromised biogenesis of sensory cilia, which identifies JS as a member of the large group of ciliopathies. Here we report on the identification of novel compound heterozygous variants (p.Y503C and p.Q485) in the centrosomal gene in a patient with JS via trio whole exome sequencing. We have studied the underlying disease mechanism in the frog , which offers fast assessment of cilia functions in a number of embryological contexts. Morpholino oligomer (MO) mediated knockdown of the orthologous gene resulted in defective mucociliary clearance in the larval epidermis, due to reduced cilia numbers and motility on multiciliated cells. To functionally assess patient alleles, mutations were analyzed in the larval skin: the p.Q485 nonsense mutation resulted in a disturbed localization of PIBF1 to the ciliary base. This mutant failed to rescue the ciliation phenotype following knockdown of endogenous . In contrast, the missense variant p.Y503C resulted in attenuated rescue capacity compared to the wild type allele. Based on these results, we conclude that in the case of this patient, JS is the result of a pathogenic combination of an amorphic and a hypomorphic allele. Our study underscores the versatility of the model to study ciliopathies such as JS in a rapid and cost-effective manner, which should render this animal model attractive for future studies of human ciliopathies.
乔伯特综合征(JS)是一种先天性常染色体隐性遗传病,在极少数情况下为X连锁遗传病。所谓的磨牙征的诊断标志描述了轴位脑扫描中中脑和后脑的形态学表现。受影响的个体表现出发育迟缓、智力残疾、共济失调、呼吸急促、睡眠呼吸暂停、眼球和舌头运动异常以及肌张力减退。在细胞水平上,JS与感觉纤毛的生物发生受损有关,这表明JS是一大类纤毛病的成员之一。在这里,我们报告通过三联全外显子测序在一名JS患者的中心体基因中鉴定出新型复合杂合变异(p.Y503C和p.Q485)。我们在青蛙中研究了潜在的疾病机制,青蛙能在许多胚胎学背景下快速评估纤毛功能。吗啉代寡聚物(MO)介导的直系同源基因敲低导致幼虫表皮的黏液纤毛清除功能缺陷,原因是多纤毛细胞上的纤毛数量减少和运动能力下降。为了在功能上评估患者的等位基因,在幼虫皮肤中分析了突变:p.Q485无义突变导致PIBF1在纤毛基部的定位紊乱。该突变体在敲低内源性基因后未能挽救纤毛形成表型。相比之下,错义变体p.Y503C与野生型等位基因相比,其挽救能力减弱。基于这些结果,我们得出结论,就该患者而言,JS是一个无义等位基因和一个低表达等位基因致病组合的结果。我们的研究强调了该模型在快速且经济高效地研究如JS等纤毛病方面的多功能性,这应该会使这种动物模型对未来人类纤毛病的研究具有吸引力。