Paul G. Allen School of Computer Science and Engineering, University of Washington, WA, USA, 98195-2350.
Nucleic Acids Res. 2019 Jun 4;47(10):e58. doi: 10.1093/nar/gkz156.
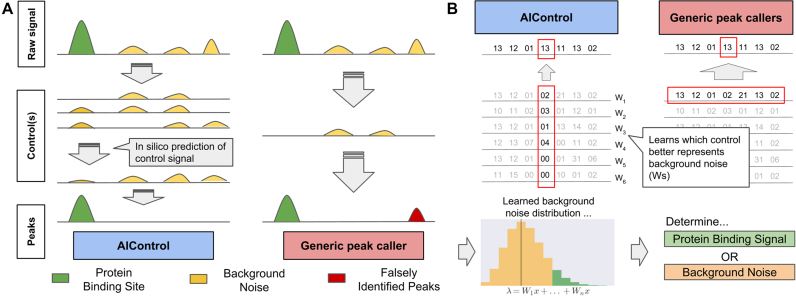
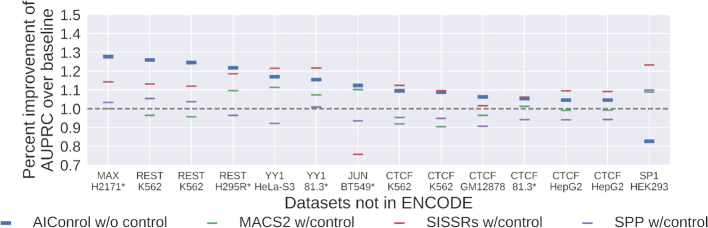
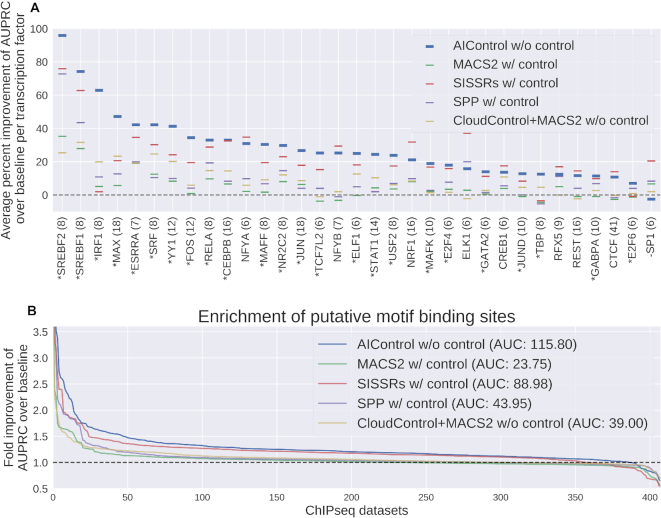
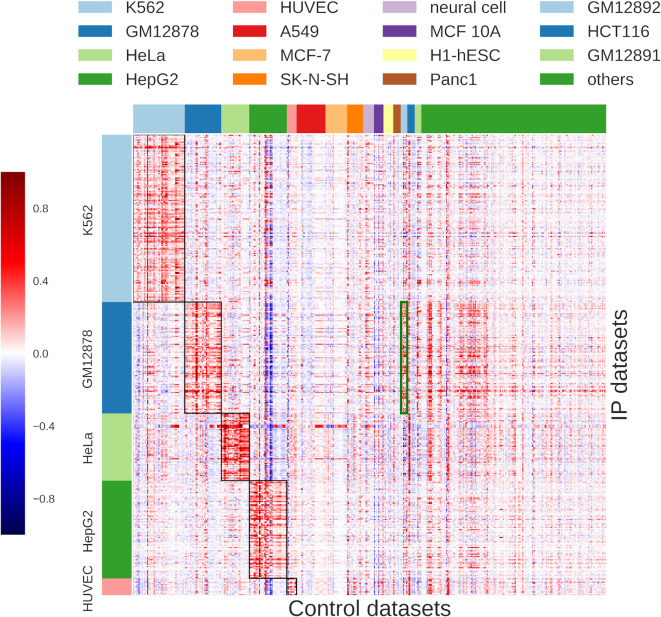
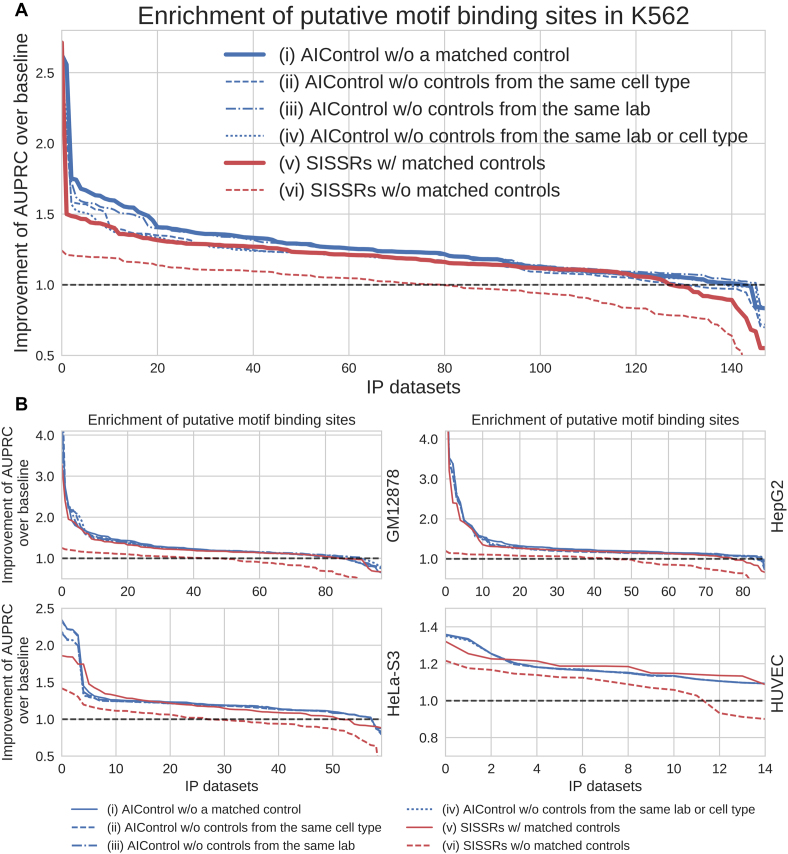
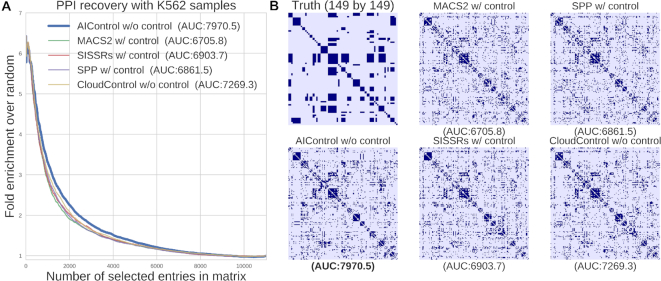
ChIP-seq is a technique to determine binding locations of transcription factors, which remains a central challenge in molecular biology. Current practice is to use a 'control' dataset to remove background signals from a immunoprecipitation (IP) 'target' dataset. We introduce the AIControl framework, which eliminates the need to obtain a control dataset and instead identifies binding peaks by estimating the distributions of background signals from many publicly available control ChIP-seq datasets. We thereby avoid the cost of running control experiments while simultaneously increasing the accuracy of binding location identification. Specifically, AIControl can (i) estimate background signals at fine resolution, (ii) systematically weigh the most appropriate control datasets in a data-driven way, (iii) capture sources of potential biases that may be missed by one control dataset and (iv) remove the need for costly and time-consuming control experiments. We applied AIControl to 410 IP datasets in the ENCODE ChIP-seq database, using 440 control datasets from 107 cell types to impute background signal. Without using matched control datasets, AIControl identified peaks that were more enriched for putative binding sites than those identified by other popular peak callers that used a matched control dataset. We also demonstrated that our framework identifies binding sites that recover documented protein interactions more accurately.
ChIP-seq 是一种用于确定转录因子结合位置的技术,这在分子生物学中仍然是一个核心挑战。目前的做法是使用“对照”数据集来去除免疫沉淀 (IP) “靶”数据集的背景信号。我们引入了 AIControl 框架,该框架无需获取对照数据集,而是通过从许多公开可用的对照 ChIP-seq 数据集中估计背景信号的分布来识别结合峰。这样,我们在避免运行对照实验成本的同时,还提高了结合位置识别的准确性。具体来说,AIControl 可以 (i) 以精细的分辨率估计背景信号,(ii) 以数据驱动的方式系统地权衡最合适的对照数据集,(iii) 捕捉可能被一个对照数据集遗漏的潜在偏差源,以及 (iv) 无需进行昂贵且耗时的对照实验。我们将 AIControl 应用于 ENCODE ChIP-seq 数据库中的 410 个 IP 数据集,使用来自 107 种细胞类型的 440 个对照数据集来估算背景信号。在不使用匹配对照数据集的情况下,AIControl 识别出的峰比其他使用匹配对照数据集的流行峰调用者识别出的峰更富集假定的结合位点。我们还证明了我们的框架可以更准确地识别出可恢复已记录蛋白相互作用的结合位点。