Wellcome Sanger Institute, Hinxton, Cambridgeshire, CB10 1SA, UK.
Department of Genetics, University of Cambridge, Downing Street, Cambridge, CB2 3EH, UK.
BMC Genomics. 2019 Mar 15;20(1):218. doi: 10.1186/s12864-019-5592-6.
Infections with helminths cause an enormous disease burden in billions of animals and plants worldwide. Large scale use of anthelmintics has driven the evolution of resistance in a number of species that infect livestock and companion animals, and there are growing concerns regarding the reduced efficacy in some human-infective helminths. Understanding the mechanisms by which resistance evolves is the focus of increasing interest; robust genetic analysis of helminths is challenging, and although many candidate genes have been proposed, the genetic basis of resistance remains poorly resolved.
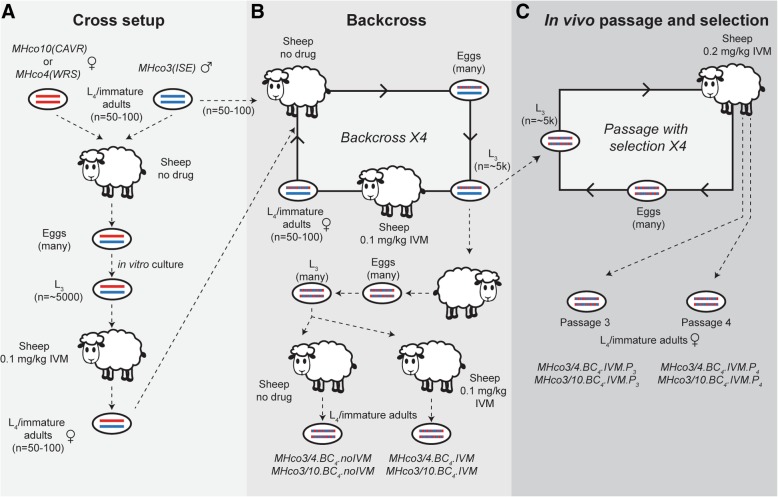
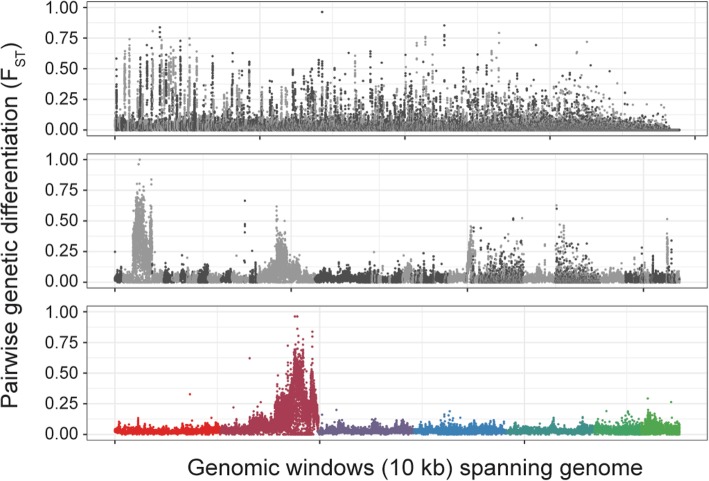
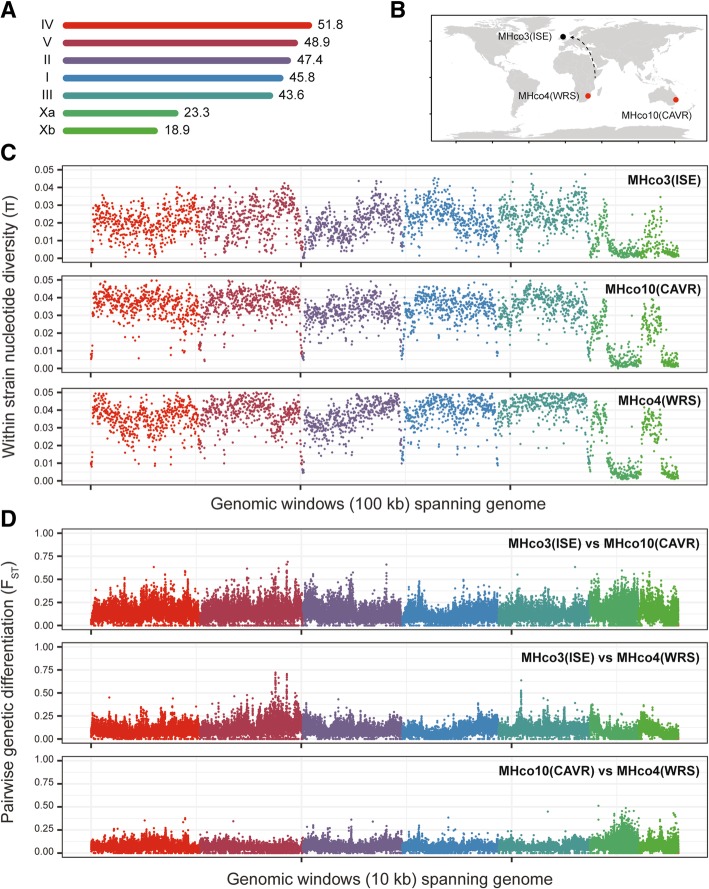
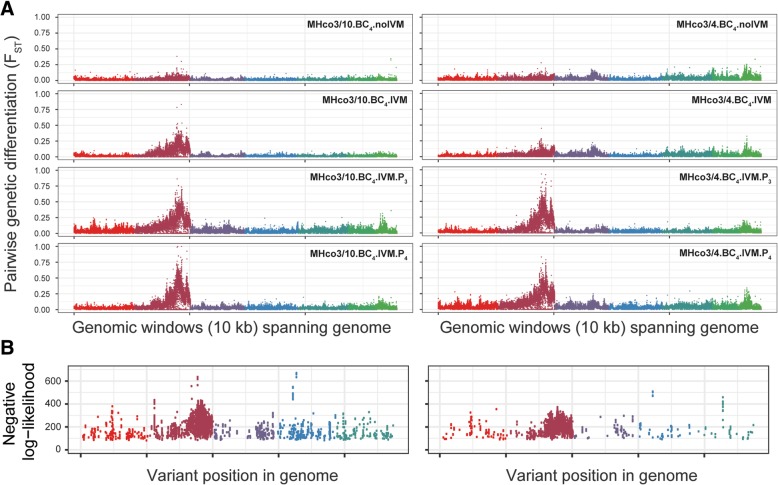
Here, we present a genome-wide analysis of two genetic crosses between ivermectin resistant and sensitive isolates of the parasitic nematode Haemonchus contortus, an economically important gastrointestinal parasite of small ruminants and a model for anthelmintic research. Whole genome sequencing of parental populations, and key stages throughout the crosses, identified extensive genomic diversity that differentiates populations, but after backcrossing and selection, a single genomic quantitative trait locus (QTL) localised on chromosome V was revealed to be associated with ivermectin resistance. This QTL was common between the two geographically and genetically divergent resistant populations and did not include any leading candidate genes, suggestive of a previously uncharacterised mechanism and/or driver of resistance. Despite limited resolution due to low recombination in this region, population genetic analyses and novel evolutionary models supported strong selection at this QTL, driven by at least partial dominance of the resistant allele, and that large resistance-associated haplotype blocks were enriched in response to selection.
We have described the genetic architecture and mode of ivermectin selection, revealing a major genomic locus associated with ivermectin resistance, the most conclusive evidence to date in any parasitic nematode. This study highlights a novel genome-wide approach to the analysis of a genetic cross in non-model organisms with extreme genetic diversity, and the importance of a high-quality reference genome in interpreting the signals of selection so identified.
寄生虫感染在全球数十亿的动植物中造成了巨大的疾病负担。在感染家畜和伴侣动物的许多物种中,大规模使用驱虫药已经导致了耐药性的产生,人们越来越担心一些人类感染性寄生虫的疗效降低。了解耐药性进化的机制是日益关注的焦点;对寄生虫进行强有力的遗传分析具有挑战性,尽管已经提出了许多候选基因,但耐药性的遗传基础仍未得到很好的解决。
在这里,我们对伊维菌素耐药和敏感的捻转血矛线虫分离株进行了全基因组分析,捻转血矛线虫是小反刍动物重要的胃肠道寄生虫,也是驱虫药研究的模型。对亲本种群和杂交过程中的关键阶段进行全基因组测序,发现了广泛的基因组多样性,可区分种群,但经过回交和选择后,一个位于第 V 号染色体上的单一基因组数量性状位点(QTL)被确定与伊维菌素耐药性相关。该 QTL 在两个地理位置和遗传上有差异的耐药种群中是共同的,且不包括任何主要的候选基因,提示存在一种以前未被描述的耐药机制和/或驱动因素。尽管由于该区域重组率低导致分辨率有限,但群体遗传分析和新的进化模型支持该 QTL 受到强烈选择,耐药等位基因至少部分显性驱动,并且在选择作用下,大的耐药相关单倍型块得到了富集。
我们描述了伊维菌素选择的遗传结构和模式,揭示了与伊维菌素耐药性相关的主要基因组位点,这是迄今为止任何寄生线虫中最确凿的证据。这项研究突出了一种新的全基因组方法,用于分析具有极端遗传多样性的非模式生物的遗传杂交,以及高质量参考基因组在解释如此确定的选择信号方面的重要性。