UMR AGAP, INRA, CIRAD, Montpellier SupAgro, Université de Montpellier, San Giuliano, France.
IRD, CIRAD, Université de Montpellier, IPME, Montpellier, France.
Ann Bot. 2019 Jul 8;123(7):1231-1251. doi: 10.1093/aob/mcz029.
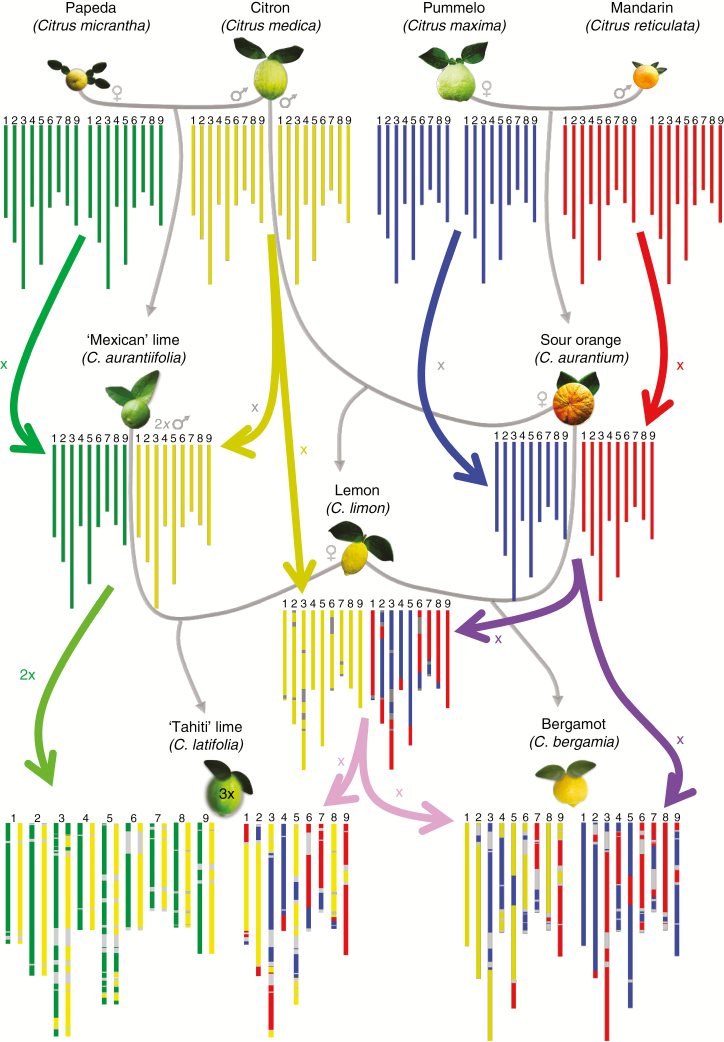
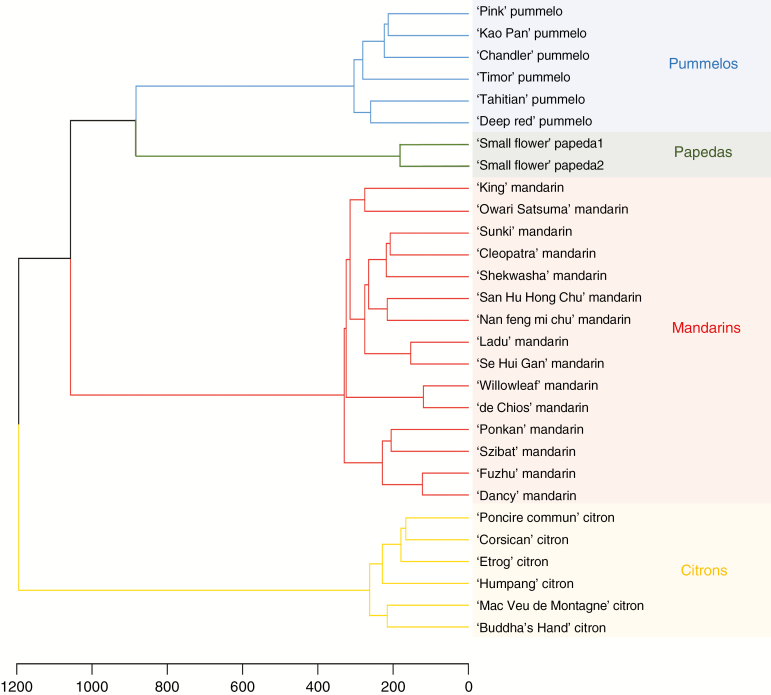
Reticulate evolution, coupled with reproductive features limiting further interspecific recombinations, results in admixed mosaics of large genomic fragments from the ancestral taxa. Whole-genome sequencing (WGS) data are powerful tools to decipher such complex genomes but still too costly to be used for large populations. The aim of this work was to develop an approach to infer phylogenomic structures in diploid, triploid and tetraploid individuals from sequencing data in reduced genome complexity libraries. The approach was applied to the cultivated Citrus gene pool resulting from reticulate evolution involving four ancestral taxa, C. maxima, C. medica, C. micrantha and C. reticulata.
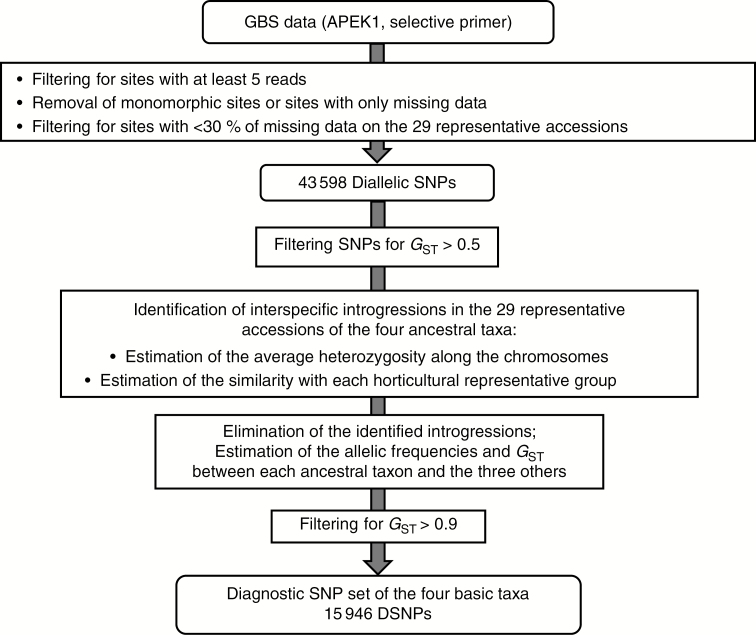
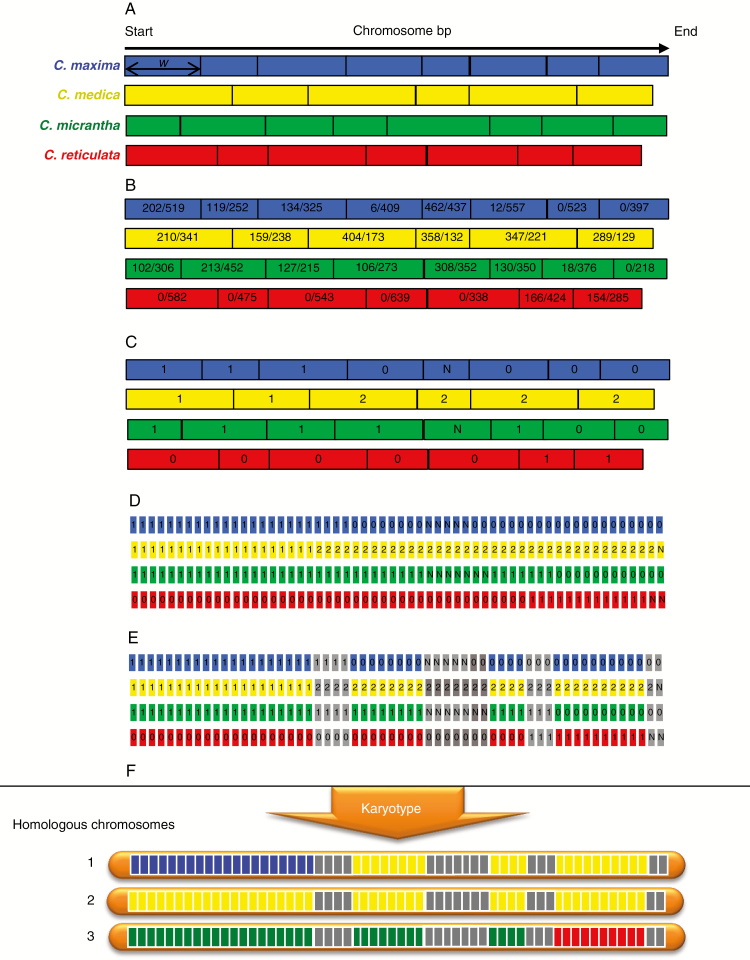
A genotyping by sequencing library was established with the restriction enzyme ApeKI applying one base (A) selection. Diagnostic single nucleotide polymorphisms (DSNPs) for the four ancestral taxa were mined in 29 representative varieties. A generic pipeline based on a maximum likelihood analysis of the number of read data was established to infer ancestral contributions along the genome of diploid, triploid and tetraploid individuals. The pipeline was applied to 48 diploid, four triploid and one tetraploid citrus accessions.
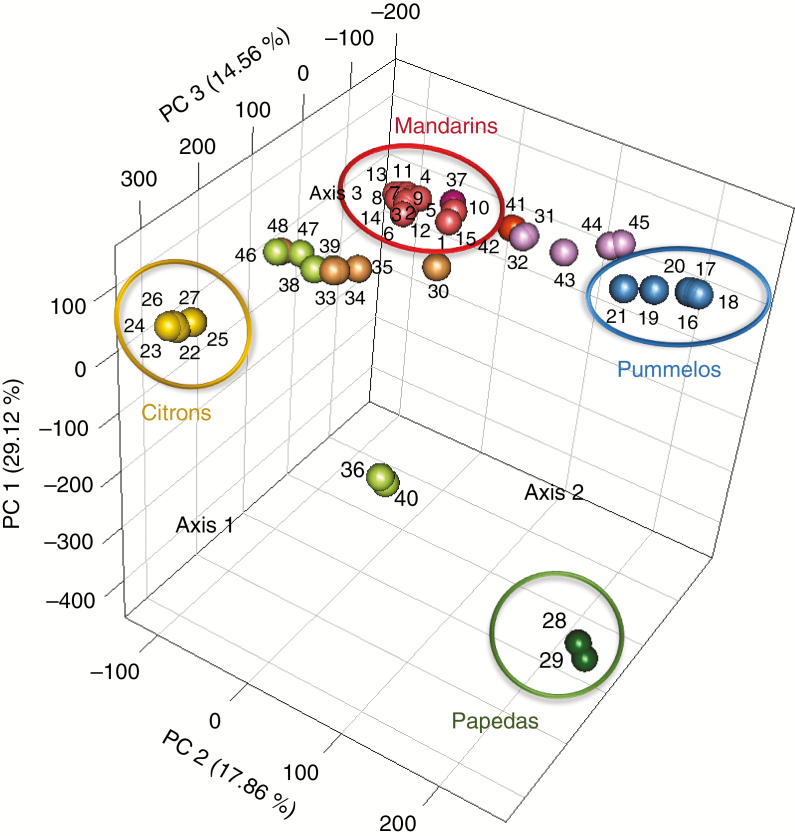
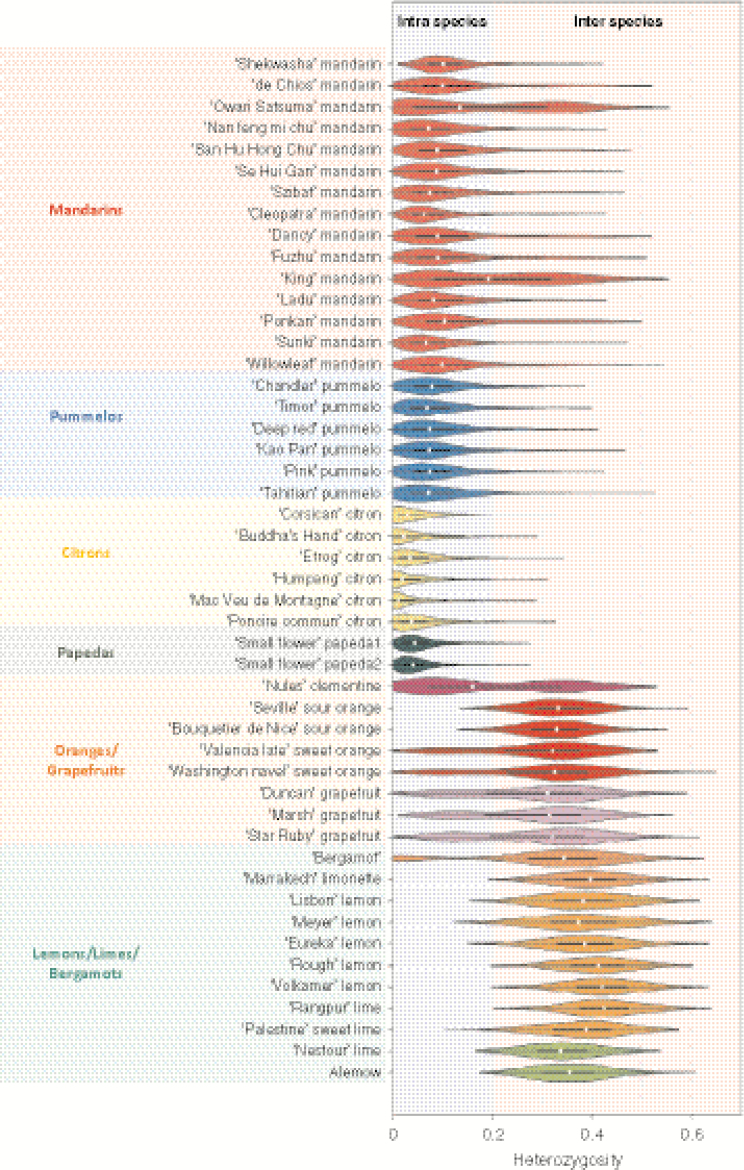
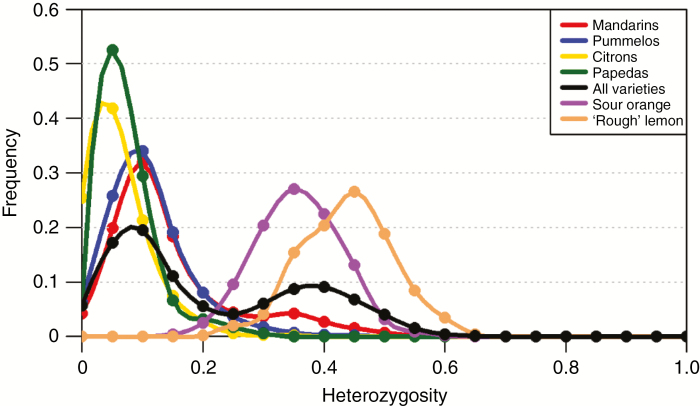
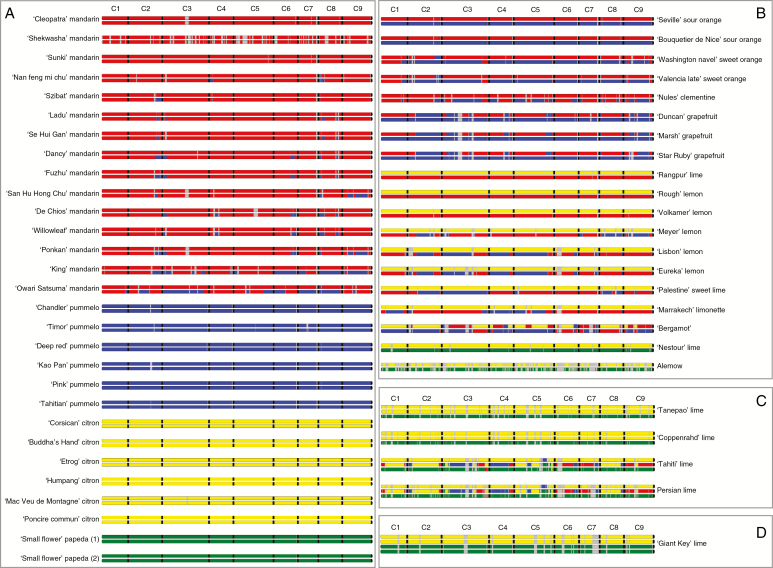
Among 43 598 mined SNPs, we identified a set of 15 946 DSNPs covering the whole genome with a distribution similar to that of gene sequences. The set efficiently inferred the phylogenomic karyotype of the 53 analysed accessions, providing patterns for common accessions very close to that previously established using WGS data. The complex phylogenomic karyotypes of 21 cultivated citrus, including bergamot, triploid and tetraploid limes, were revealed for the first time.
The pipeline, available online, efficiently inferred the phylogenomic structures of diploid, triploid and tetraploid citrus. It will be useful for any species whose reproductive behaviour resulted in an interspecific mosaic of large genomic fragments. It can also be used for the first generations of interspecific breeding schemes.
网状进化,加上限制进一步种间重组的生殖特征,导致来自祖先分类群的大基因组片段的混合镶嵌体。全基因组测序 (WGS) 数据是破译此类复杂基因组的有力工具,但仍然过于昂贵,无法用于大群体。本研究的目的是开发一种方法,用于从简化基因组复杂度文库的测序数据中推断二倍体、三倍体和四倍体个体的系统基因组结构。该方法应用于涉及四个祖先分类群(C. maxima、C. medica、C. micrantha 和 C. reticulata)的网状进化产生的栽培柑橘基因库。
使用限制性内切酶 ApeKI 建立了一个测序文库,该酶应用了一个碱基(A)选择。在 29 个代表品种中挖掘了用于四个祖先分类群的诊断单核苷酸多态性 (DSNP)。建立了一个基于最大似然分析读数据数量的通用管道,用于推断二倍体、三倍体和四倍体个体基因组中祖先的贡献。该管道应用于 48 个二倍体、四个三倍体和一个四倍体柑橘品种。
在挖掘的 43598 个 SNP 中,我们确定了一组 15946 个 DSNPs,覆盖整个基因组,其分布与基因序列相似。该组有效地推断了 53 个分析品种的系统基因组核型,提供的模式与以前使用 WGS 数据建立的模式非常相似。首次揭示了 21 个栽培柑橘,包括佛手柑、三倍体和四倍体酸橙的复杂系统基因组核型。
该管道可在线使用,有效地推断了二倍体、三倍体和四倍体柑橘的系统基因组结构。它将对任何因种间大基因组片段镶嵌而导致生殖行为复杂的物种有用。它还可用于种间杂交计划的第一代。