Department of Pharmacology , Penn State University College of Medicine , Hershey , Pennsylvania 17033 , United States.
Department of Biochemistry & Molecular Biology , Penn State University College of Medicine , Hershey , Pennsylvania 17033 , United States.
J Chem Inf Model. 2019 Jun 24;59(6):2509-2515. doi: 10.1021/acs.jcim.8b00905. Epub 2019 Apr 17.
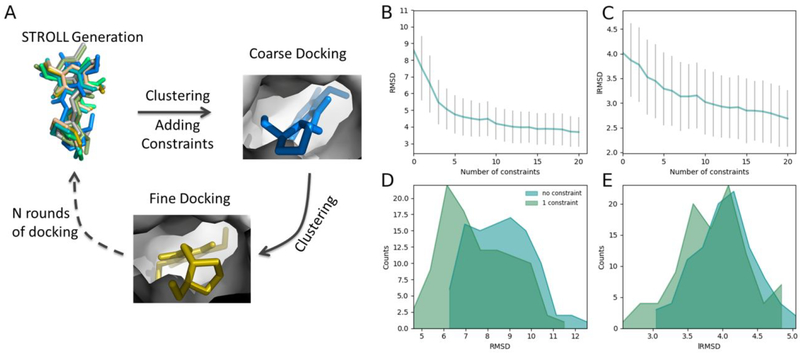
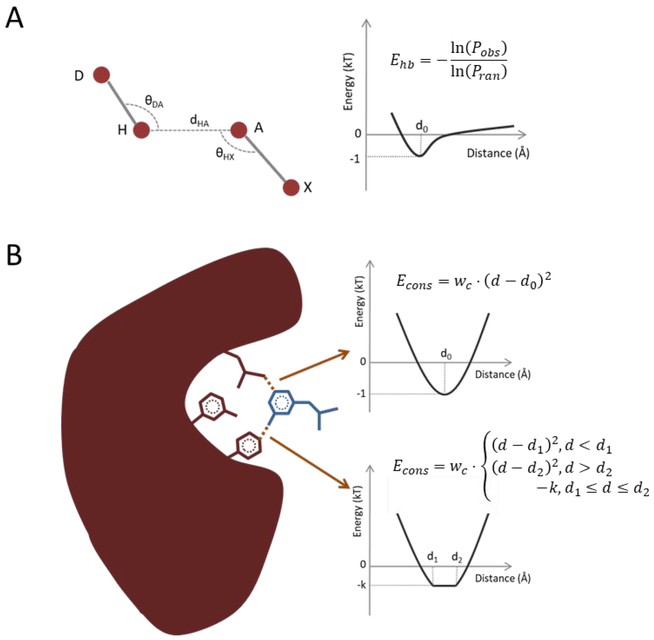
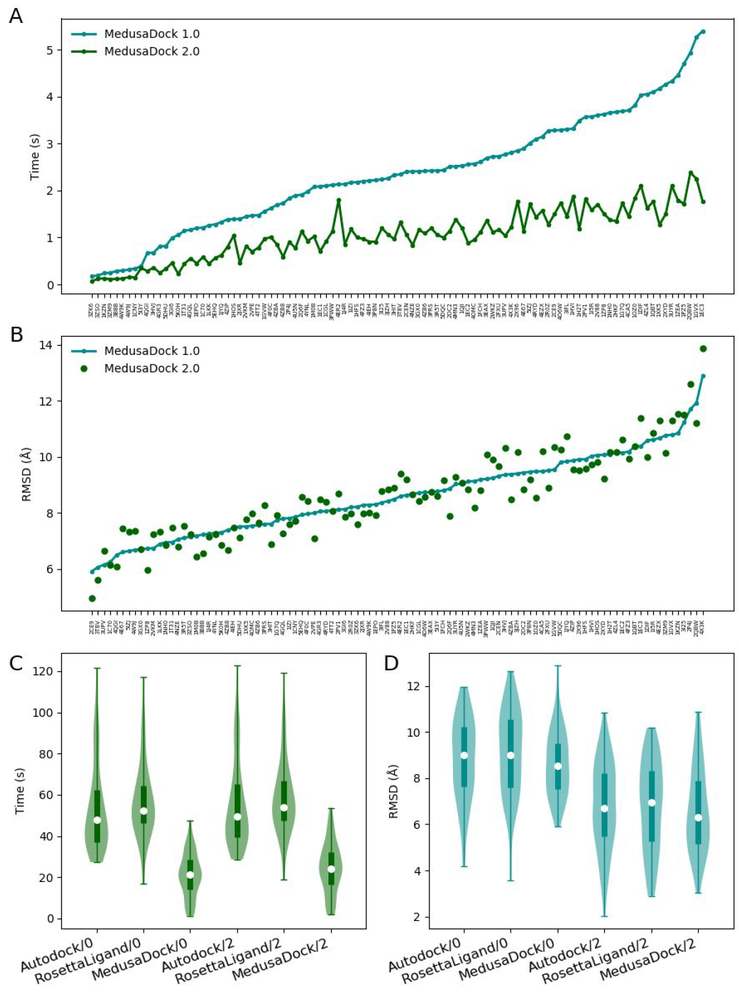
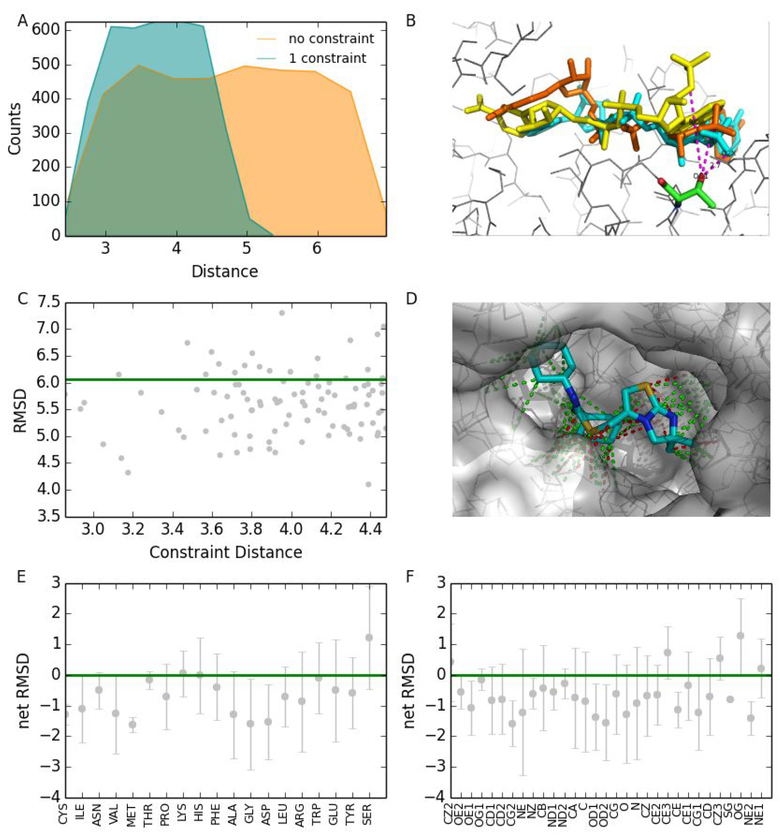
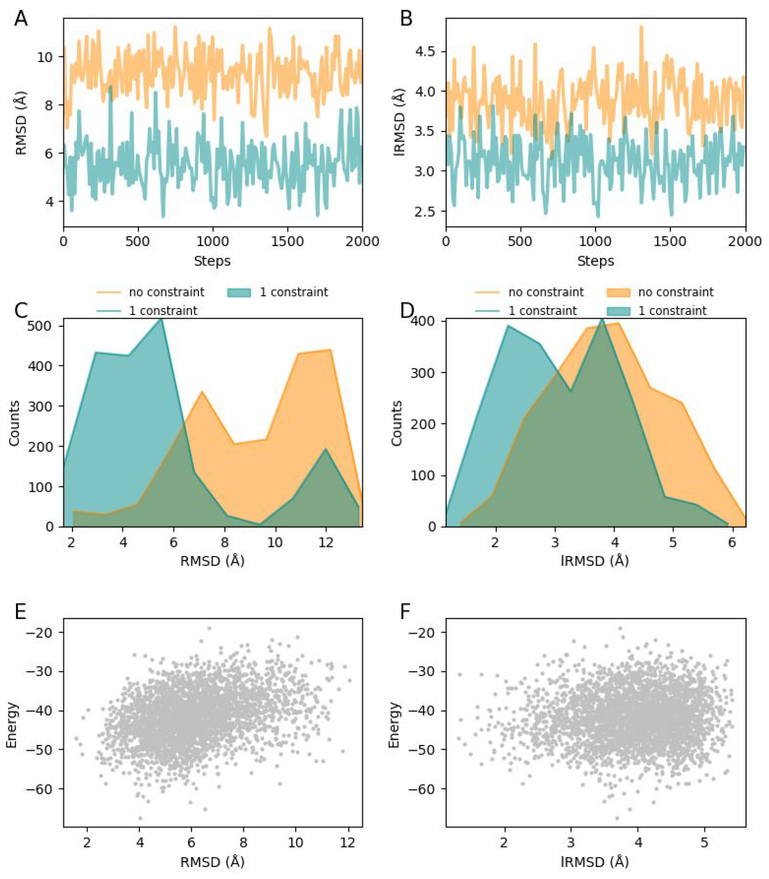
Molecular docking is the key ingredient of virtual drug screening, a promising and cost-effective approach for finding new drugs. A critical limitation of this approach is the inadequate sampling efficiency of both ligand and/or receptor conformations for finding the lowest energy bound state. To circumvent this limitation, we develop a protein-ligand docking methodology capable of incorporating structural constraints, experimentally derived or theoretically predicted, to improve accuracy and efficiency. We develop a web server with a user-friendly online graphical interface as a platform for accurate and efficient protein-ligand molecule docking.
分子对接是虚拟药物筛选的关键组成部分,是一种有前途且具有成本效益的寻找新药的方法。这种方法的一个关键限制是配体和/或受体构象的采样效率不足,无法找到最低能量结合态。为了克服这一限制,我们开发了一种能够结合结构约束的蛋白质-配体对接方法,这些约束可以是实验得出或理论预测的,以提高准确性和效率。我们开发了一个带有用户友好的在线图形界面的网络服务器,作为准确和高效的蛋白质-配体分子对接的平台。