Forli Stefano, Huey Ruth, Pique Michael E, Sanner Michel F, Goodsell David S, Olson Arthur J
Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, California, USA.
Nat Protoc. 2016 May;11(5):905-19. doi: 10.1038/nprot.2016.051. Epub 2016 Apr 14.
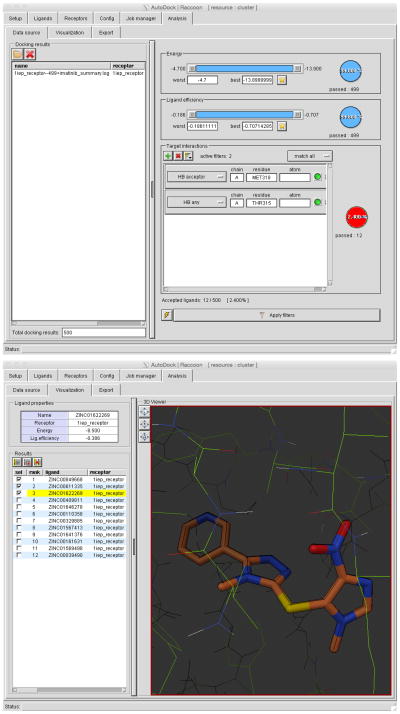
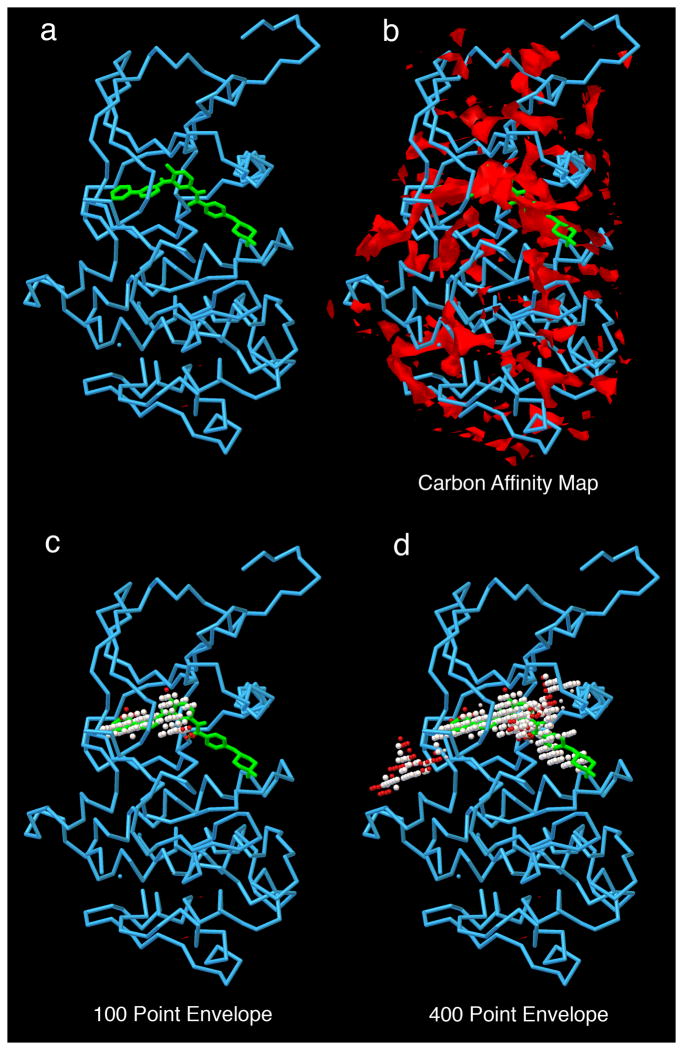
Computational docking can be used to predict bound conformations and free energies of binding for small-molecule ligands to macromolecular targets. Docking is widely used for the study of biomolecular interactions and mechanisms, and it is applied to structure-based drug design. The methods are fast enough to allow virtual screening of ligand libraries containing tens of thousands of compounds. This protocol covers the docking and virtual screening methods provided by the AutoDock suite of programs, including a basic docking of a drug molecule with an anticancer target, a virtual screen of this target with a small ligand library, docking with selective receptor flexibility, active site prediction and docking with explicit hydration. The entire protocol will require ∼5 h.
计算对接可用于预测小分子配体与大分子靶标的结合构象和结合自由能。对接广泛应用于生物分子相互作用和机制的研究,并应用于基于结构的药物设计。这些方法速度足够快,能够对包含数以万计化合物的配体库进行虚拟筛选。本方案涵盖了AutoDock程序套件提供的对接和虚拟筛选方法,包括药物分子与抗癌靶标的基本对接、用小型配体库对该靶标进行虚拟筛选、具有选择性受体灵活性的对接、活性位点预测以及显式水合对接。整个方案大约需要5小时。