Laboratory of DNA Damage Signaling, Chiba Cancer Center Research Institute, Japan.
Laboratory of Oncogenomics, Chiba Cancer Center Research Institute, Japan.
FEBS Open Bio. 2019 May;9(5):935-946. doi: 10.1002/2211-5463.12636. Epub 2019 Apr 18.
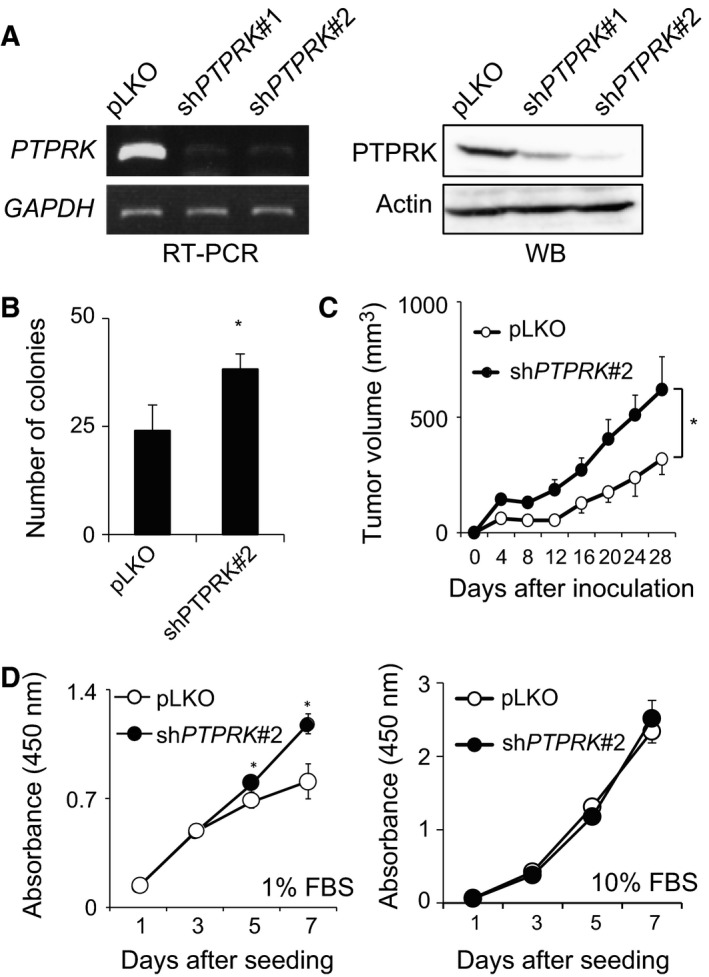
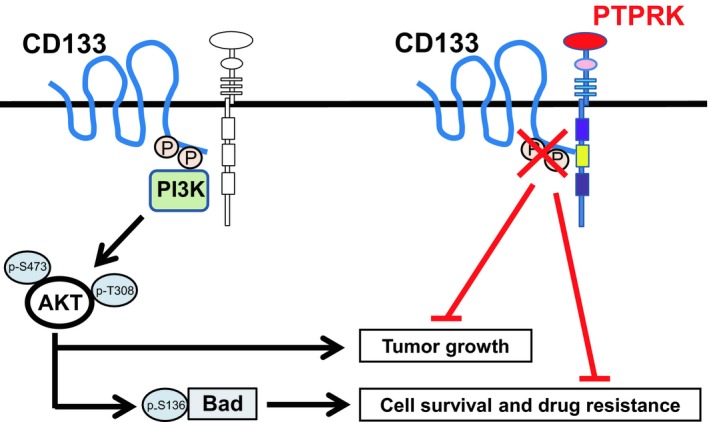
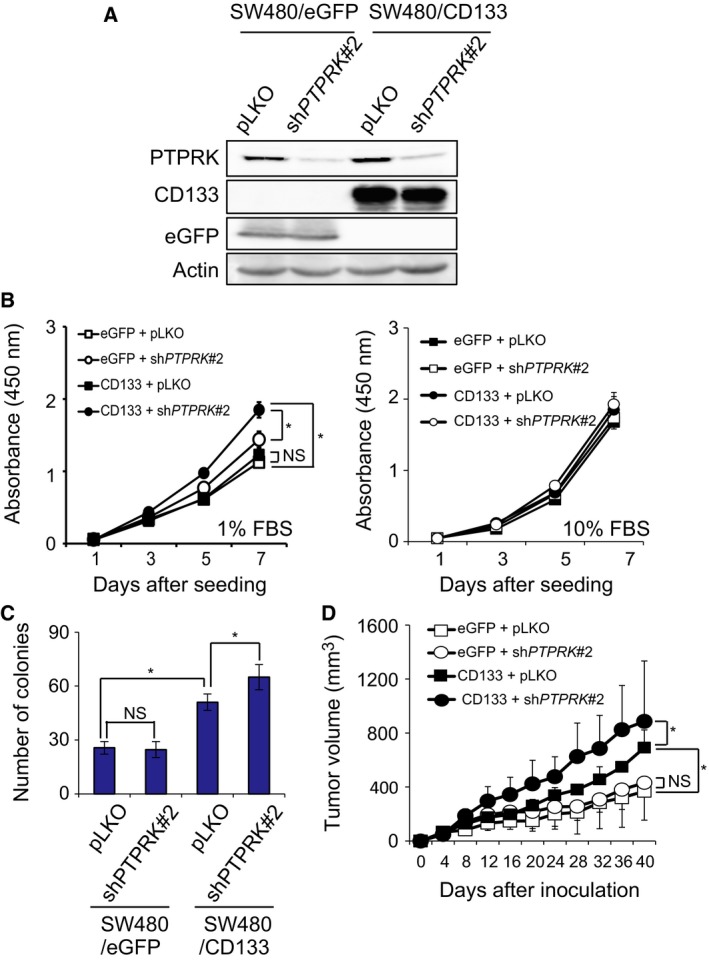
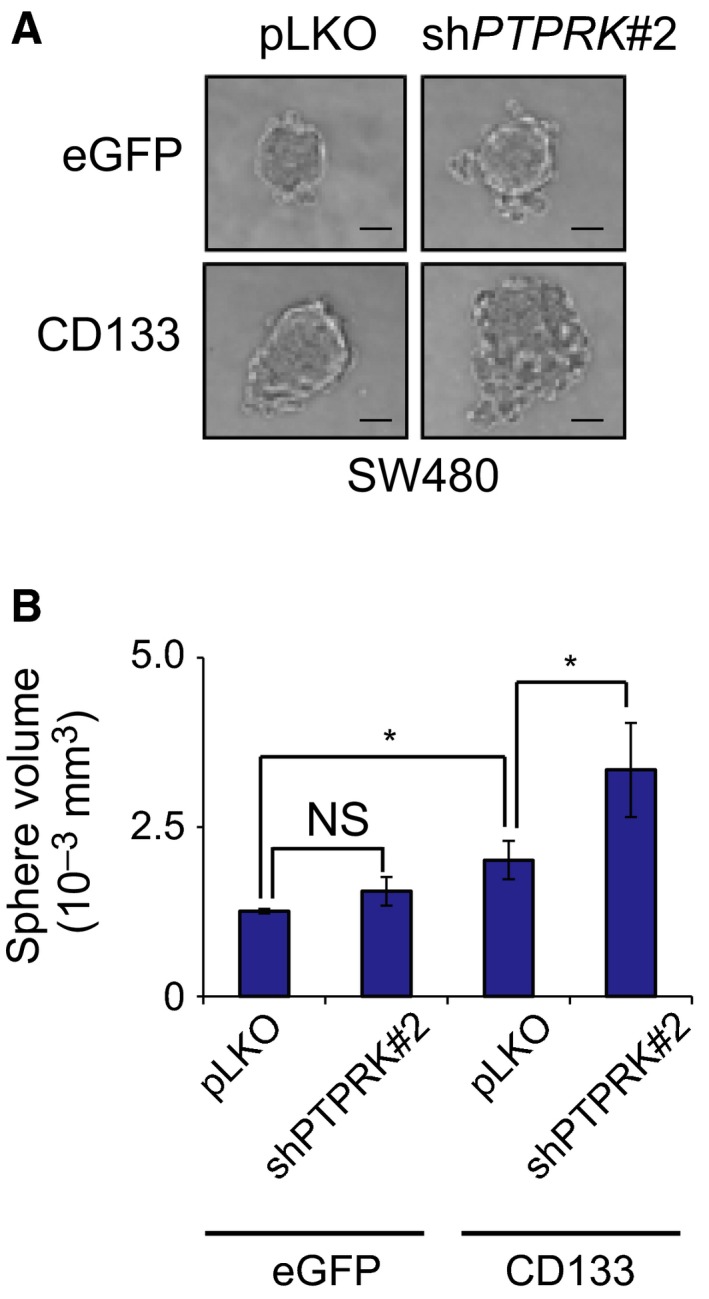
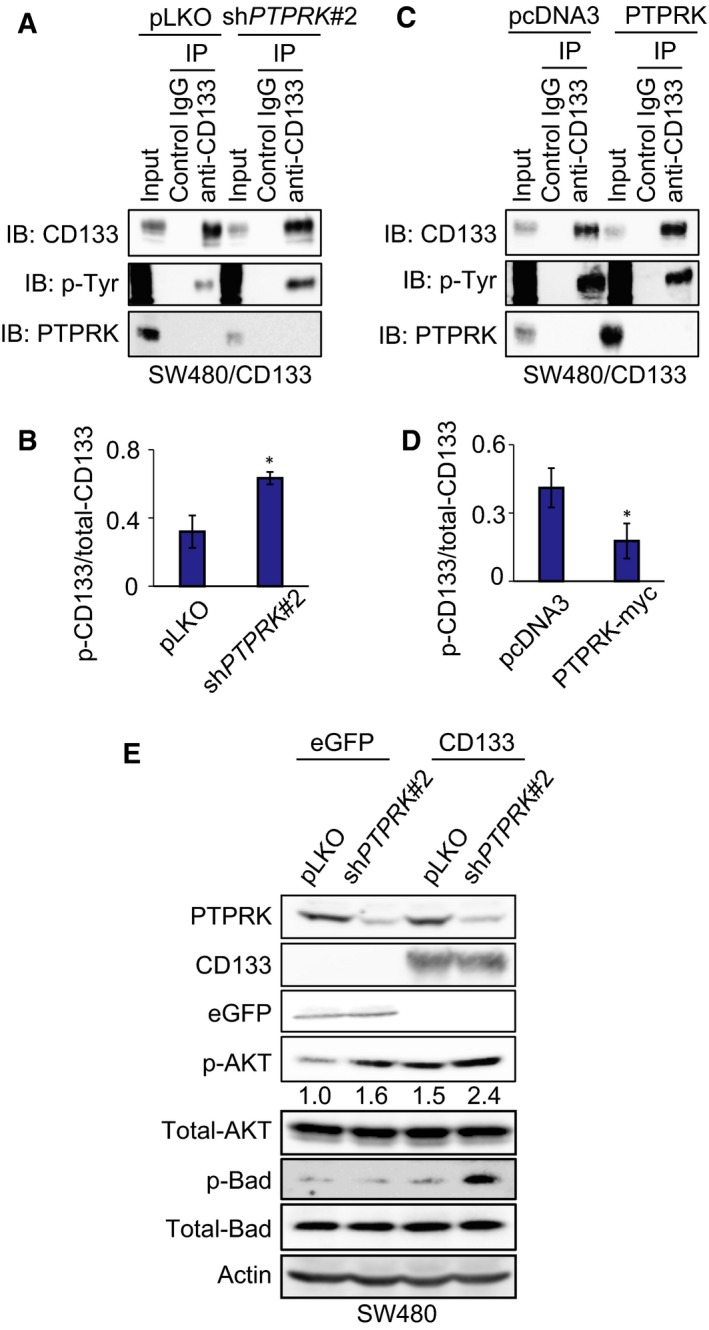
Receptor-type protein tyrosine phosphatase κ (PTPRK) is considered to be a candidate tumor suppressor. PTPRK dephosphorylates CD133, which is a stem cell marker; phosphorylated CD133 accelerates xenograft tumor growth of colon cancer cells through the activation of AKT, but the functional significance of this has remained elusive. In this study, we have demonstrated that knockdown of PTPRK potentiates the pro-oncogenic CD133-AKT pathway in colon cancer cells. Intriguingly, depletion of PTPRK significantly reduced sensitivity to the anti-cancer drug oxaliplatin and was accompanied by up-regulation of phosphorylation of Bad, a downstream target of AKT. Together, our present observations strongly suggest that the CD133-PTPRK axis plays a pivotal role in the regulation of colon cancer progression as well as drug resistance.
受体型蛋白酪氨酸磷酸酶 κ(PTPRK)被认为是一种候选肿瘤抑制因子。PTPRK 去磷酸化 CD133,CD133 是一种干细胞标记物;磷酸化的 CD133 通过激活 AKT 加速结肠癌细胞的异种移植肿瘤生长,但这一功能意义仍不清楚。在这项研究中,我们已经证明,PTPRK 的敲低增强了结肠癌细胞中致癌性的 CD133-AKT 通路。有趣的是,PTPRK 的耗竭显著降低了对抗癌药物奥沙利铂的敏感性,并伴随着 AKT 的下游靶标 Bad 的磷酸化上调。总之,我们目前的观察结果强烈表明,CD133-PTPRK 轴在调节结肠癌的进展以及耐药性方面起着关键作用。