Department of Ophthalmology, Donders Institute for Brain, Cognition and Behavior, Radboud University Medical Center, Nijmegen, The Netherlands.
Department of Human Genetics, Donders Institute for Brain, Cognition and Behavior, Radboud University Medical Center, Nijmegen, The Netherlands.
Mol Genet Genomic Med. 2019 Jun;7(6):e660. doi: 10.1002/mgg3.660. Epub 2019 Apr 4.
Early-onset photoreceptor dystrophies are a major cause of irreversible visual impairment in children and young adults. This clinically heterogeneous group of disorders can be caused by mutations in many genes. Nevertheless, to date, 30%-40% of cases remain genetically unexplained. In view of expanding therapeutic options, it is essential to obtain a molecular diagnosis in these patients as well. In this study, we aimed to identify the genetic cause in two siblings with genetically unexplained retinal disease.
Whole exome sequencing was performed to identify the causative variants in two siblings in whom a single pathogenic variant in TULP1 was found previously. Patients were clinically evaluated, including assessment of the medical history, slit-lamp biomicroscopy, and ophthalmoscopy. In addition, a functional analysis of the putative splice variant in TULP1 was performed using a midigene assay.
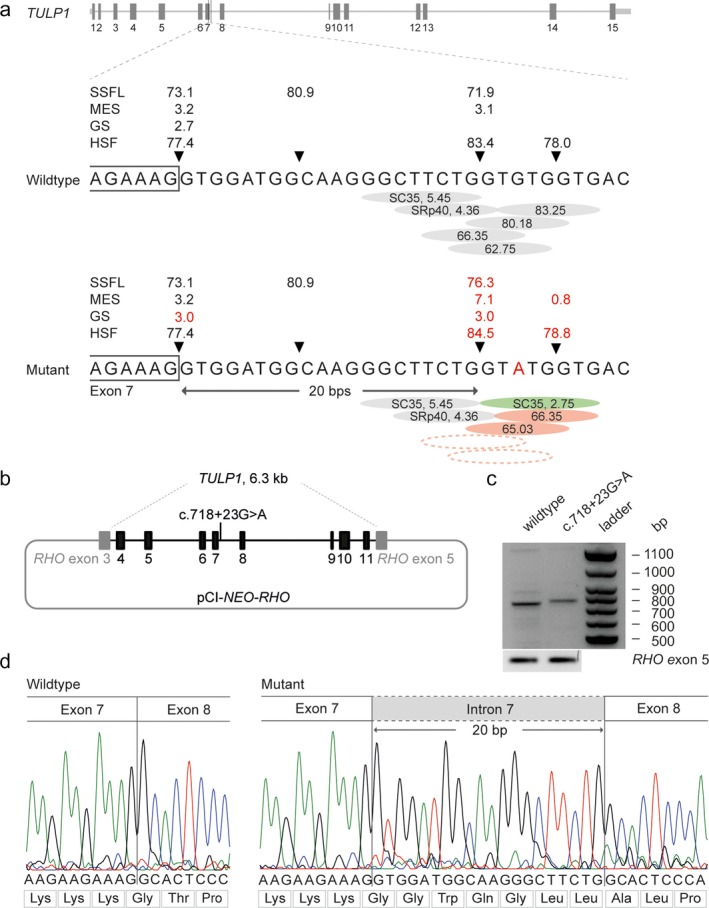
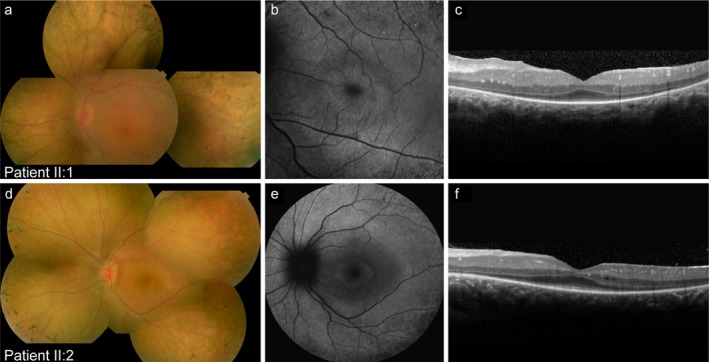
Clinical assessment showed a typical early-onset photoreceptor dystrophy in both the patients. Whole exome sequencing identified two pathogenic variants in TULP1, a c.1445G>A (p.(Arg482Gln)) missense mutation and an intronic c.718+23G>A variant. Segregation analysis confirmed that both siblings were compound heterozygous for the TULP1 c.718+23G>A and c.1445G>A variants, while the unaffected parents were heterozygous. The midigene assay for the c.718+23G>A variant confirmed an elongation of exon 7 leading to a frameshift.
Here, we report the first near-exon RNA splice variant that is not present in a consensus splice site sequence in TULP1, which was found in a compound heterozygous manner with a previously described pathogenic TULP1 variant in two patients with an early-onset photoreceptor dystrophy. We provide proof of pathogenicity for this splice variant by performing an in vitro midigene splice assay, and highlight the importance of analysis of noncoding regions beyond the noncanonical splice sites in patients with inherited retinal diseases.
早发性光感受器营养不良是儿童和青年不可逆视力损害的主要原因。这组临床异质性疾病可能由许多基因突变引起。然而,迄今为止,仍有 30%-40%的病例未得到遗传解释。鉴于治疗选择的不断扩大,在这些患者中获得分子诊断也至关重要。在这项研究中,我们旨在确定两名遗传上未解释的视网膜疾病患者的遗传原因。
对两名先前发现 TULP1 中存在单一致病性变异的兄弟姐妹进行全外显子组测序,以鉴定致病变异。对患者进行临床评估,包括评估病史、裂隙灯生物显微镜检查和眼底检查。此外,还使用 midigene 测定法对 TULP1 中的假定剪接变体进行了功能分析。
临床评估显示两名患者均存在典型的早发性光感受器营养不良。全外显子组测序在 TULP1 中发现了两个致病性变异,c.1445G>A(p.(Arg482Gln))错义突变和内含子 c.718+23G>A 变异。连锁分析证实,两名兄弟姐妹均为 TULP1 c.718+23G>A 和 c.1445G>A 变异的复合杂合子,而未受影响的父母则为杂合子。c.718+23G>A 变异的 midigene 测定证实外显子 7 的延长导致移码。
在这里,我们报告了 TULP1 中第一个不在保守剪接位点序列中的近外显子 RNA 剪接变体,该变体以复合杂合方式存在于两名早发性光感受器营养不良患者中,与先前描述的致病性 TULP1 变异一起存在。我们通过进行体外 midigene 剪接测定,为该剪接变体提供了致病性证据,并强调了在遗传性视网膜疾病患者中分析非编码区域的重要性,超出了非规范剪接位点。