Michael Smith Laboratories, University of British Columbia, Vancouver, BC, Canada, V6T 1Z4.
Michael Smith Genome Sciences Centre, BC Cancer, Vancouver, BC, Canada, V5Z 4S6.
Nat Commun. 2019 Apr 5;10(1):1556. doi: 10.1038/s41467-019-09583-2.
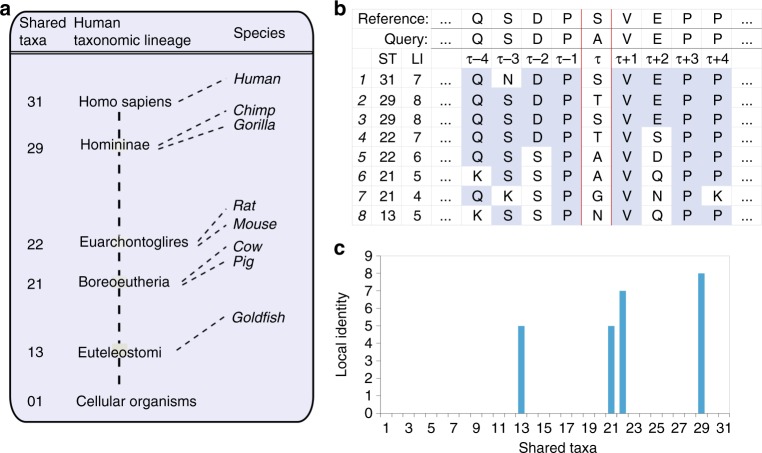
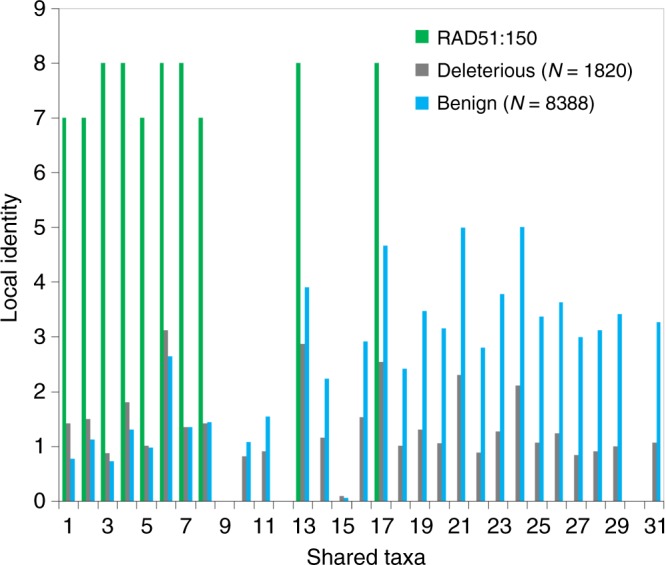
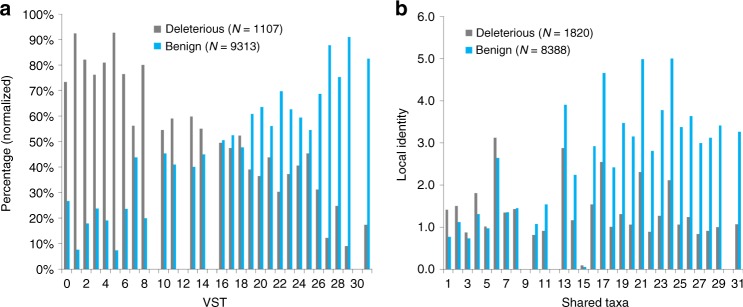
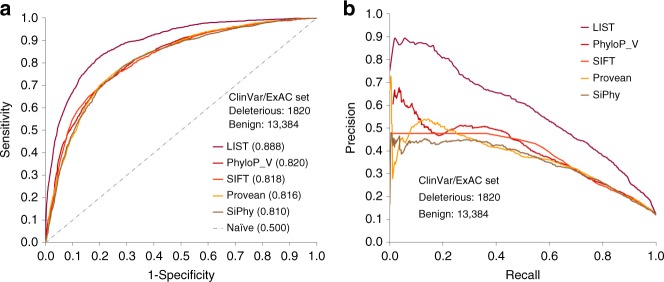
Selective pressures on protein-coding regions that provide fitness advantages can lead to the regions' fixation and conservation in genome duplications and speciation events. Consequently, conservation analyses relying on sequence similarities are exploited by a myriad of applications across all biosciences to identify functionally important protein regions. While very potent, existing conservation measures based on multiple sequence alignments are so pervasive that improvements to solutions of many problems have become incremental. We introduce a new framework for evolutionary conservation with measures that exploit taxonomy distances across species. Results show that our taxonomy-based framework comfortably outperforms existing conservation measures in identifying deleterious variants observed in the human population, including variants located in non-abundant sequence domains such as intrinsically disordered regions. The predictive power of our approach emphasizes that the phenotypic effects of sequence variants can be taxonomy-level specific and thus, conservation needs to be interpreted accordingly.
对提供适应优势的蛋白质编码区域的选择压力可导致这些区域在基因组加倍和物种形成事件中固定和保守。因此,基于序列相似性的保守性分析被广泛应用于所有生物科学领域,以识别功能重要的蛋白质区域。虽然现有的基于多重序列比对的保守性度量方法非常有效,但它们已经如此普遍,以至于许多问题的解决方案的改进都变得微不足道。我们引入了一种新的进化保守性框架,该框架利用了跨物种的分类学距离来进行度量。结果表明,我们的基于分类学的框架在识别人类群体中观察到的有害变异方面表现出色,包括位于非丰富序列域(如无序区域)中的变异。我们的方法的预测能力强调了序列变异的表型效应可能具有分类水平特异性,因此需要相应地解释保守性。