Lou Weiyang, Ding Bisha, Xu Liang, Fan Weimin
Program of Innovative Cancer Therapeutics, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, Key Laboratory of Organ Transplantation, First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China.
Key Laboratory of Organ Transplantation, Hangzhou, China.
Front Mol Neurosci. 2019 Mar 26;12:66. doi: 10.3389/fnmol.2019.00066. eCollection 2019.
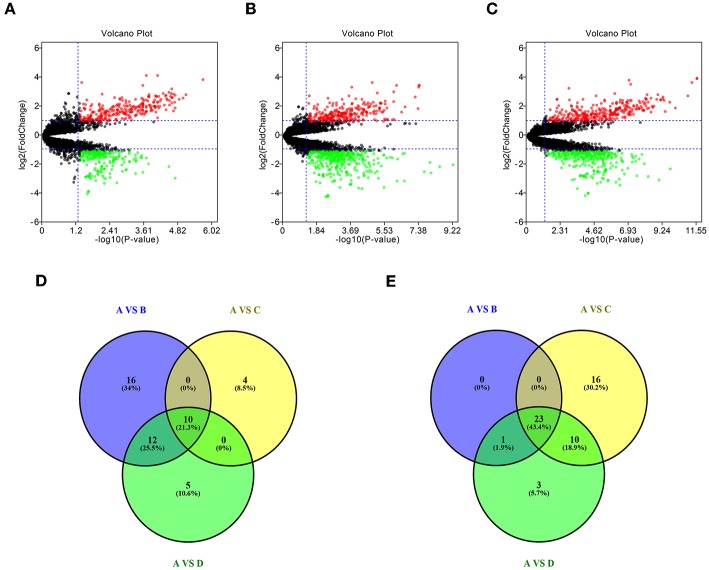
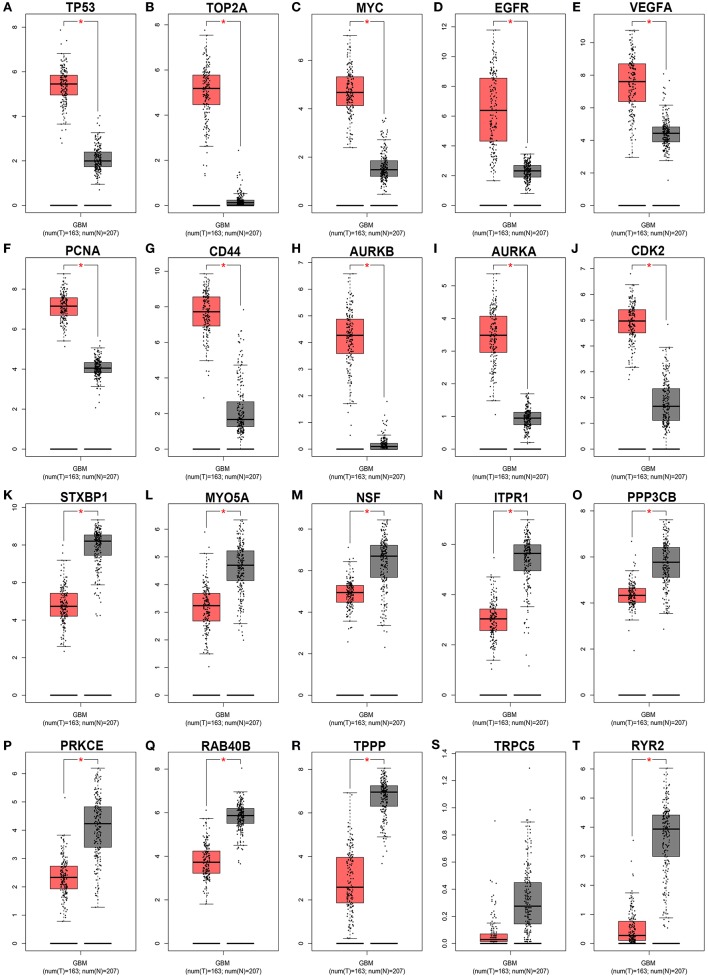
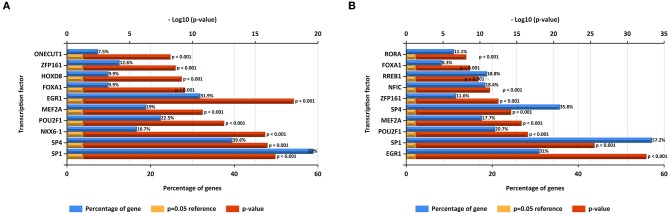
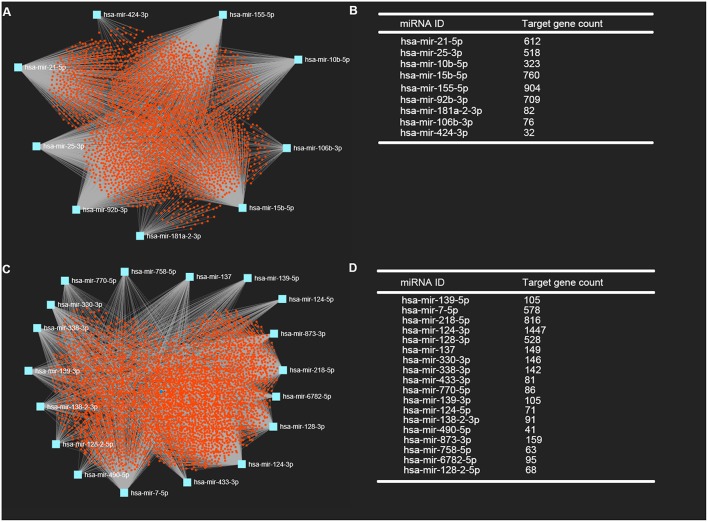
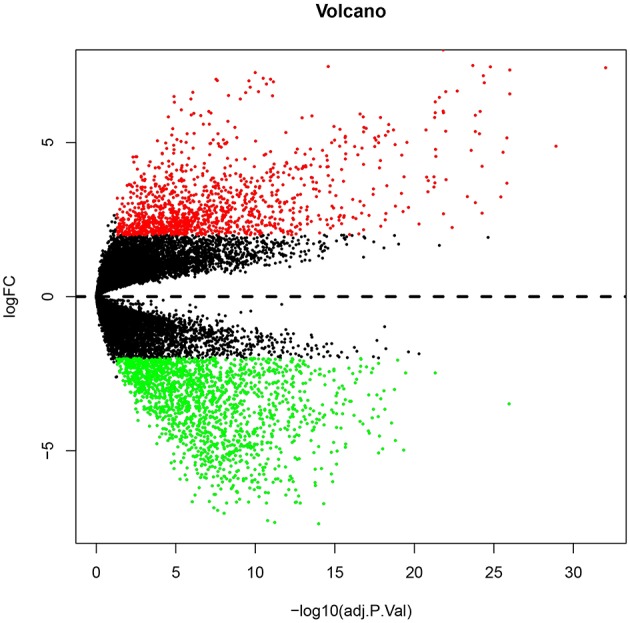
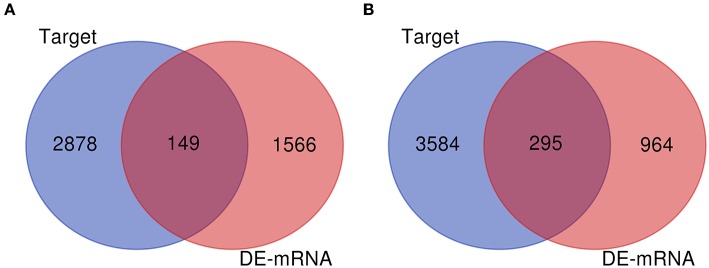
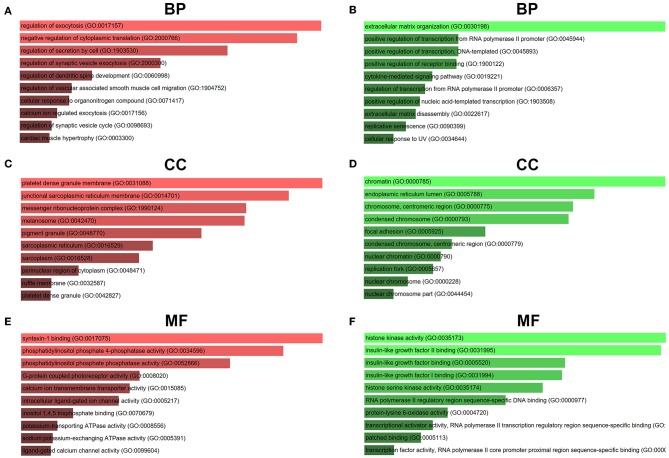
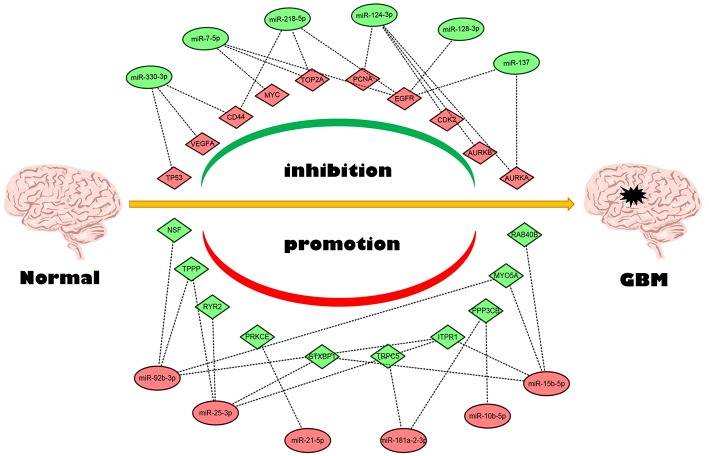
Glioblastoma multiforme (GBM), the most common and aggressive human malignant brain tumor, is notorious for its limited treatment options and poor prognosis. MicroRNAs (miRNAs) are found to be involved in tumorigenesis of GBM. However, a comprehensive miRNA-mRNA regulatory network has still not been established. A miRNA microarray dataset (GSE90603) was obtained from GEO database. Then, we employed GEO2R tool to perform differential expression analysis. Potential transcription factors and target genes of screened differentially expressed miRNAs (DE-miRNAs) were predicted. The GBM mRNA dataset were downloaded from TCGA database for identifying differentially expressed genes (DEGs). Next, GO annotation and KEGG pathway enrichment analysis was conducted. PPI network was then established, and hub genes were identified via Cytoscape software. The expression and prognostic roles of hub genes was further evaluated. Total 33 DE-miRNAs, consisting of 10 upregulated DE-miRNAs and 23 downregulated DE-miRNAs, were screened. SP1 was predicted to potentially regulate most of screened DE-miRNAs. Three thousand and twenty seven and 3,879 predicted target genes were obtained for upregulated and downregulated DE-miRNAs, respectively. Subsequently, 1,715 upregulated DEGs and 1,259 downregulated DEGs were identified. Then, 149 and 295 potential downregulated and upregulated genes commonly appeared in target genes of DE-miRNAs and DEGs were selected for GO annotation and KEGG pathway enrichment analysis. The downregulated genes were significantly enriched in cGMP-PKG signaling pathway and calcium signaling pathway whereas the upregulated genes were enriched in pathways in cancer and PI3K-Akt signaling pathway. Construction and analysis of PPI network showed that STXBP1 and TP53 were recognized as hub genes with the highest connectivity degrees. Expression analytic result of the top 20 hub genes in GBM using GEPIA database was generally identical with previous differential expression analysis for TCGA data. EGFR, PPP3CB, and MYO5A expression was significantly associated with patients' OS. In this study, we established a potential GBM-related miRNA-mRNA regulatory network, which explores a comprehensive understanding of the molecular mechanisms and provides key clues in seeking novel therapeutic targets for GBM. In the future, more experiments need to be performed to validate our current findings.
多形性胶质母细胞瘤(GBM)是最常见且侵袭性最强的人类恶性脑肿瘤,因其治疗选择有限且预后较差而声名狼藉。研究发现,微小RNA(miRNA)参与了GBM的肿瘤发生过程。然而,一个全面的miRNA-信使核糖核酸(mRNA)调控网络仍未建立。从基因表达综合数据库(GEO)数据库中获取了一个miRNA微阵列数据集(GSE90603)。然后,我们使用GEO2R工具进行差异表达分析。对筛选出的差异表达miRNA(DE-miRNA)的潜在转录因子和靶基因进行了预测。从癌症基因组图谱(TCGA)数据库下载GBM的mRNA数据集,以鉴定差异表达基因(DEG)。接下来,进行基因本体(GO)注释和京都基因与基因组百科全书(KEGG)通路富集分析。随后建立了蛋白质-蛋白质相互作用(PPI)网络,并通过Cytoscape软件鉴定了枢纽基因。进一步评估了枢纽基因的表达和预后作用。共筛选出33个DE-miRNA,其中包括10个上调的DE-miRNA和23个下调的DE-miRNA。预测SP1可能调控大多数筛选出的DE-miRNA。上调和下调的DE-miRNA分别获得了3027个和3879个预测靶基因。随后,鉴定出1715个上调的DEG和1259个下调的DEG。然后,从DE-miRNA和DEG的靶基因中共同出现的149个潜在下调基因和295个潜在上调基因中进行选择,用于GO注释和KEGG通路富集分析。下调基因在环磷酸鸟苷-蛋白激酶G(cGMP-PKG)信号通路和钙信号通路中显著富集,而上调基因则在癌症相关通路和磷脂酰肌醇-3激酶-蛋白激酶B(PI3K-Akt)信号通路中富集。PPI网络的构建和分析表明, syntaxin binding protein 1(STXBP1)和肿瘤蛋白p53(TP53)被识别为连接度最高的枢纽基因。使用GEPIA数据库对GBM中前20个枢纽基因的表达分析结果与之前对TCGA数据的差异表达分析基本一致。表皮生长因子受体(EGFR)、蛋白磷酸酶2B(PPP3CB)和肌球蛋白ⅤA(MYO5A)的表达与患者的总生存期(OS)显著相关。在本研究中,我们建立了一个潜在的GBM相关miRNA-mRNA调控网络,该网络有助于全面了解分子机制,并为寻找GBM的新型治疗靶点提供关键线索。未来,需要进行更多实验来验证我们目前的发现。