Fumarola Claudia, Bozza Nicole, Castelli Riccardo, Ferlenghi Francesca, Marseglia Giuseppe, Lodola Alessio, Bonelli Mara, La Monica Silvia, Cretella Daniele, Alfieri Roberta, Minari Roberta, Galetti Maricla, Tiseo Marcello, Ardizzoni Andrea, Mor Marco, Petronini Pier Giorgio
Department of Medicine and Surgery, University of Parma, Parma, Italy.
Department of Food and Drug, University of Parma, Parma, Italy.
Front Oncol. 2019 Mar 26;9:179. doi: 10.3389/fonc.2019.00179. eCollection 2019.
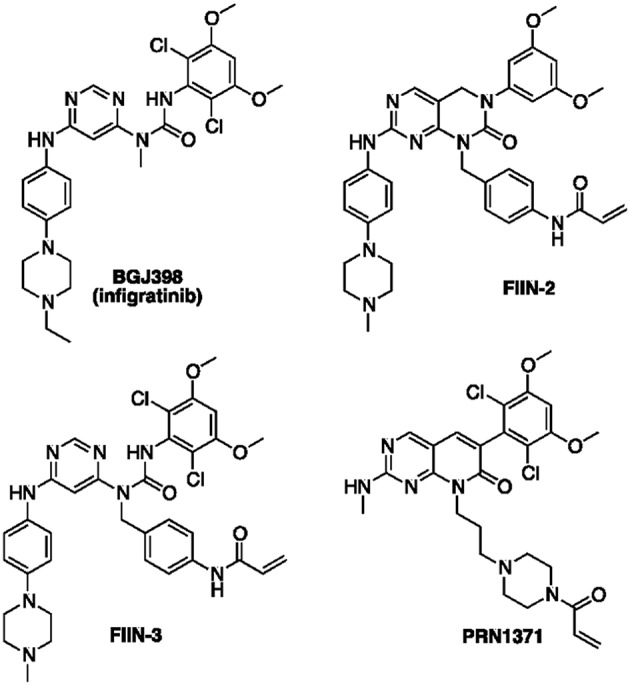
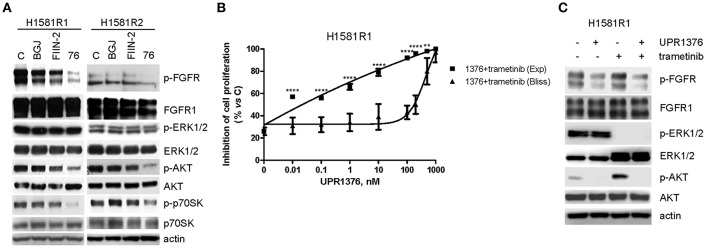
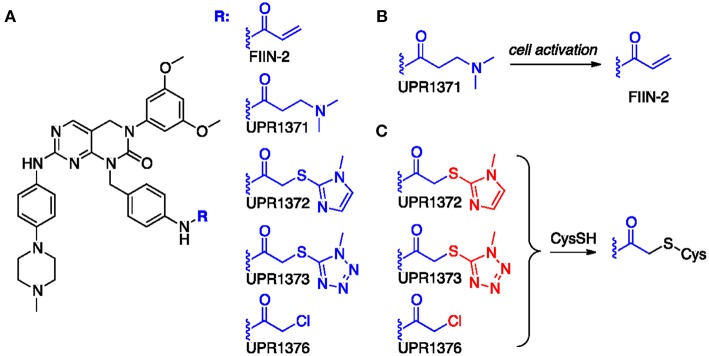
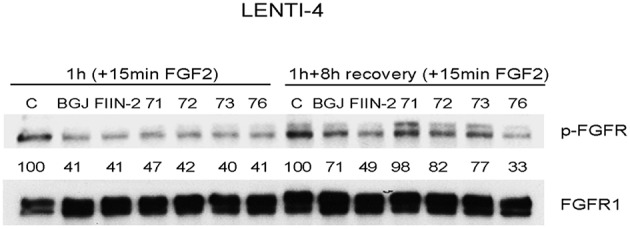
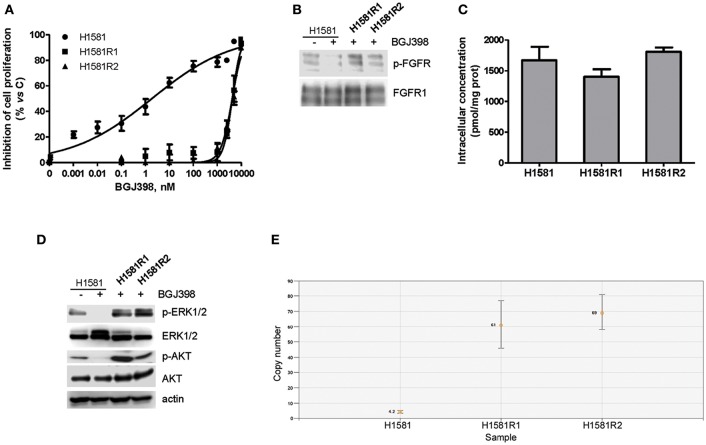
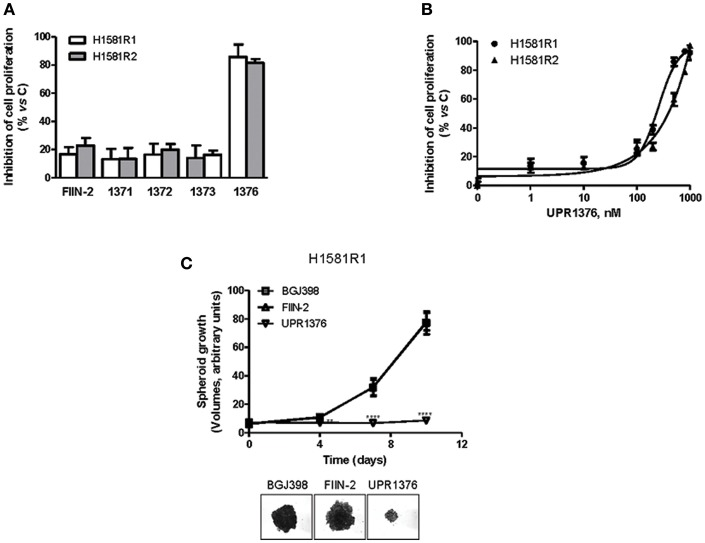
Fibroblast Growth Factor Receptors (FGFR1-4) have a critical role in the progression of several human cancers, including Squamous Non-Small-Cell Lung Cancer (SQCLC). Both non-selective and selective reversible FGFR inhibitors are under clinical investigation for the treatment of patients with tumors harboring FGFR alterations. Despite their potential efficacy, the clinical development of these drugs has encountered several challenges, including toxicity, and the appearance of drug resistance. Recent efforts have been directed at development of irreversible FGFR inhibitors, which have the potential to exert superior anti-proliferative activity in tumors carrying FGFR alterations. With this in mind, we synthetized, and investigated a set of novel inhibitors possessing a warhead potentially able to covalently bind a cysteine in the P-loop of FGFR. Among them, the chloroacetamide UPR1376 resulted able to irreversible inhibit FGFR1 phosphorylation in FGFR1 over-expressing cells generated from SQCLC SKMES-1 cells. In addition, this compound inhibited cell proliferation in FGFR1-amplified H1581 cells with a potency higher than the reversible inhibitor BGJ398 (infigratinib), while sparing FGFR1 low-expressing cells. The anti-proliferative effects of UPR1376 were demonstrated in both 2D and 3D systems and were associated with the inhibition of MAPK and AKT/mTOR signaling pathways. UPR1376 inhibited cell proliferation also in two BGJ398-resistant cell clones generated from H1581 by chronic exposure to BGJ398, although at concentrations higher than those effective in the parental cells, likely due to the persistent activation of the MAPK pathway associated to amplification. Combined blockade of FGFR1 and MAPK signaling, by UPR1376 and trametinib respectively, significantly enhanced the efficacy of UPR1376, providing a means of circumventing resistance to FGFR1 inhibition. Our findings suggest that the insertion of a chloroacetamide warhead on a suitable scaffold, as exemplified by UPR1376, is a valuable strategy to develop a novel generation of FGFR inhibitors for the treatment of SQCLC patients with FGFR alterations.
成纤维细胞生长因子受体(FGFR1 - 4)在包括鳞状非小细胞肺癌(SQCLC)在内的多种人类癌症进展中起关键作用。非选择性和选择性可逆FGFR抑制剂都正在进行临床试验,用于治疗携带FGFR改变的肿瘤患者。尽管它们具有潜在疗效,但这些药物的临床开发遇到了几个挑战,包括毒性和耐药性的出现。最近的努力致力于开发不可逆FGFR抑制剂,其有可能在携带FGFR改变的肿瘤中发挥更强的抗增殖活性。考虑到这一点,我们合成并研究了一组新型抑制剂,它们具有一个弹头,有可能与FGFR的P环中的半胱氨酸共价结合。其中,氯乙酰胺UPR1376能够不可逆地抑制由SQCLC SKMES - 1细胞产生的FGFR1过表达细胞中的FGFR1磷酸化。此外,该化合物在FGFR1扩增的H1581细胞中抑制细胞增殖的效力高于可逆抑制剂BGJ398(英菲格拉替尼),同时对FGFR1低表达细胞无影响。UPR1376的抗增殖作用在二维和三维系统中均得到证实,并且与MAPK和AKT/mTOR信号通路的抑制有关。UPR1376在通过长期暴露于BGJ398从H1581产生的两个BGJ398耐药细胞克隆中也抑制细胞增殖,尽管所需浓度高于对亲本细胞有效的浓度,这可能是由于与扩增相关的MAPK通路持续激活所致。分别用UPR1376和曲美替尼联合阻断FGFR1和MAPK信号,显著增强了UPR1376的疗效,提供了一种规避对FGFR1抑制耐药的方法。我们的研究结果表明,如UPR1376所示,在合适的支架上插入氯乙酰胺弹头是开发新一代用于治疗携带FGFR改变的SQCLC患者的FGFR抑制剂的有价值策略。