Institute of Analytical Chemistry and Radiochemistry, Leopold-Franzens University, Innrain 80/82, CCB-Center for Chemistry and Biomedicine, 6020-Innsbruck, Austria.
Faculty of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland.
Molecules. 2019 Apr 10;24(7):1402. doi: 10.3390/molecules24071402.
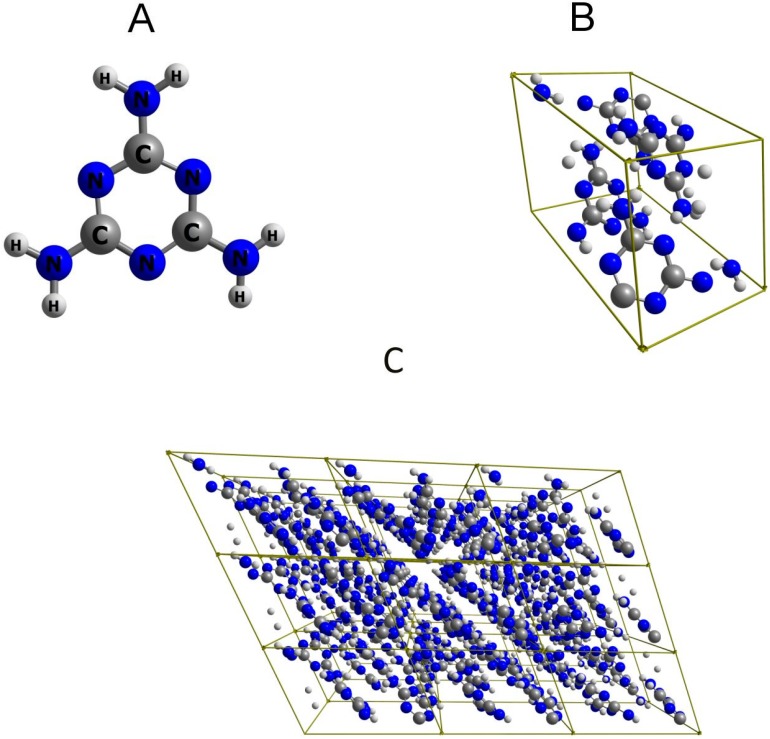
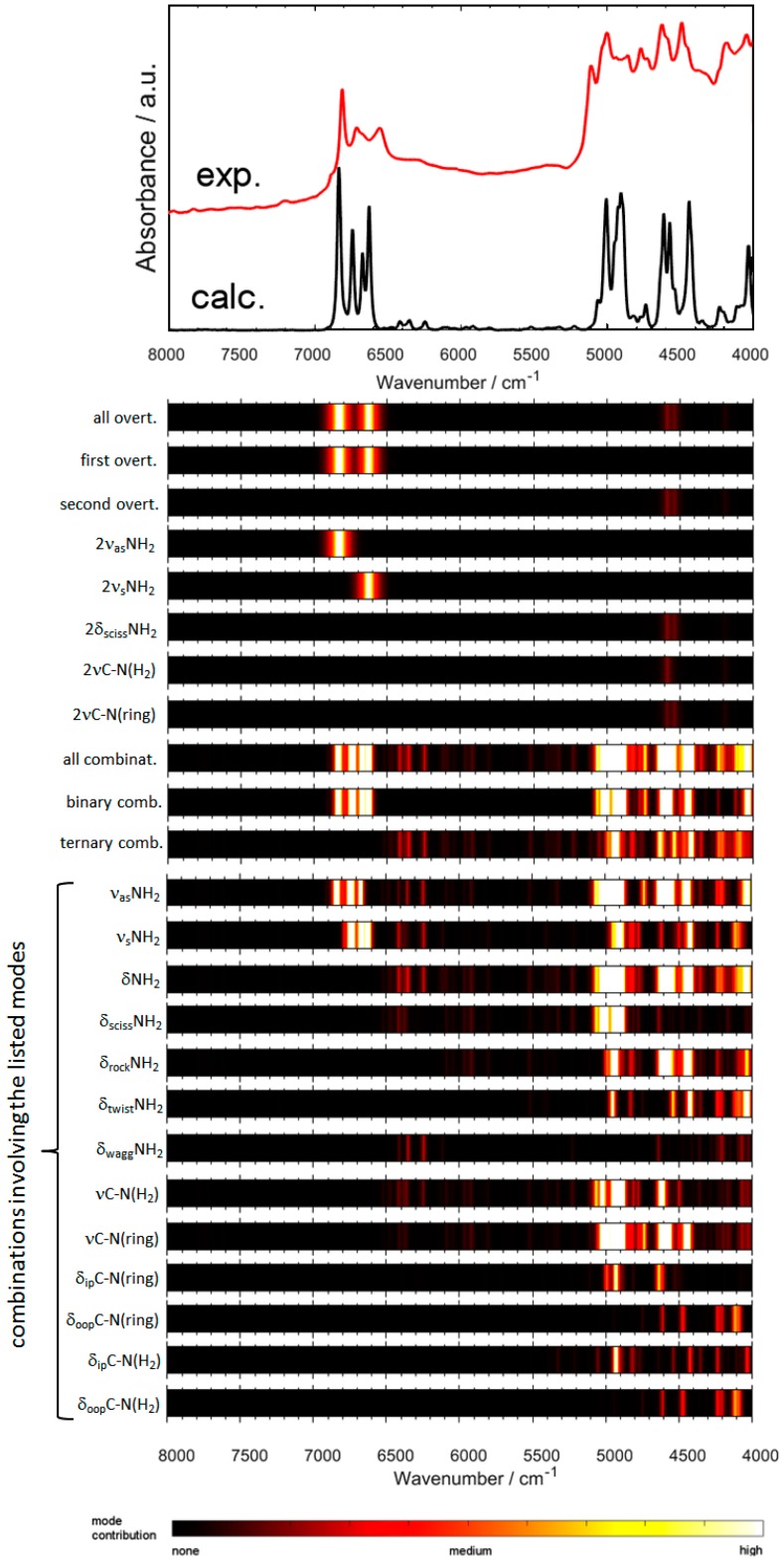
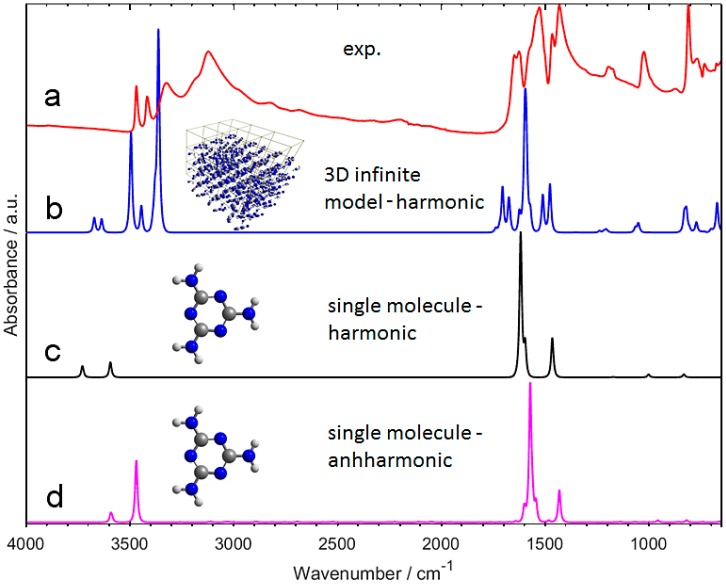
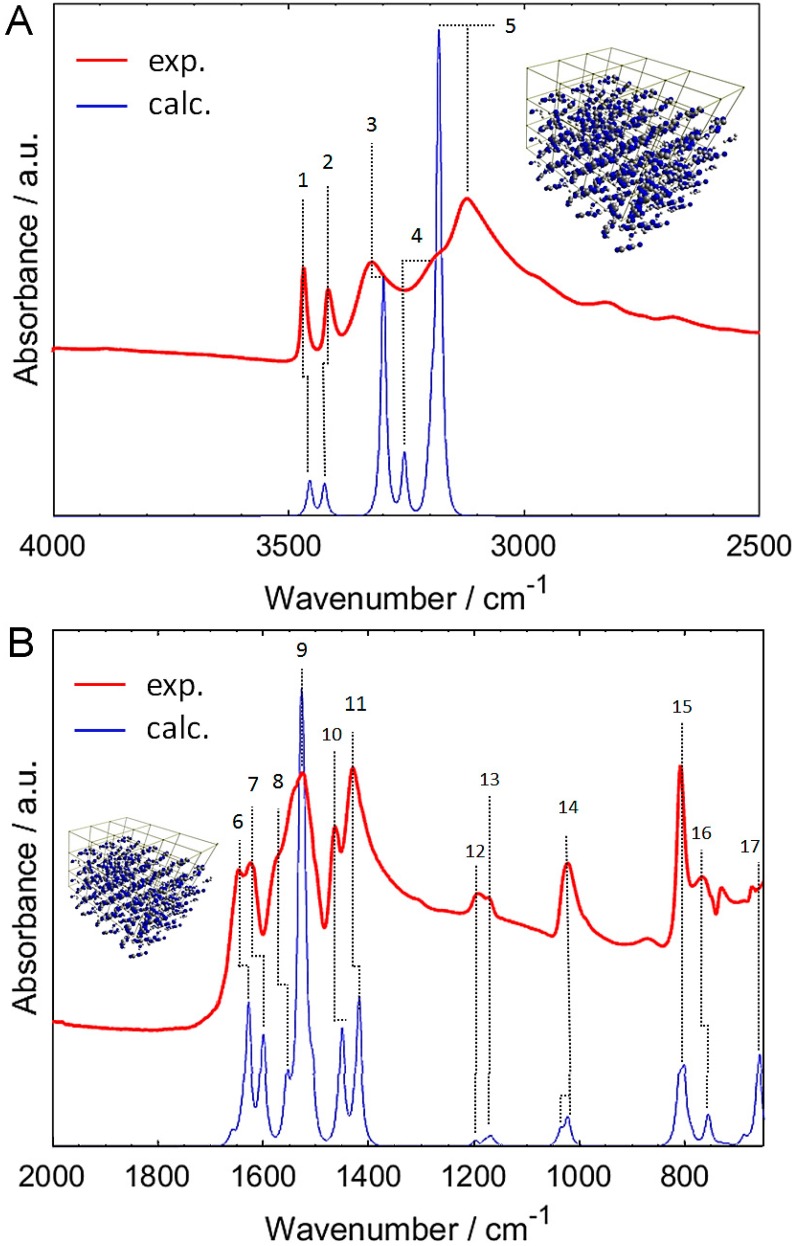
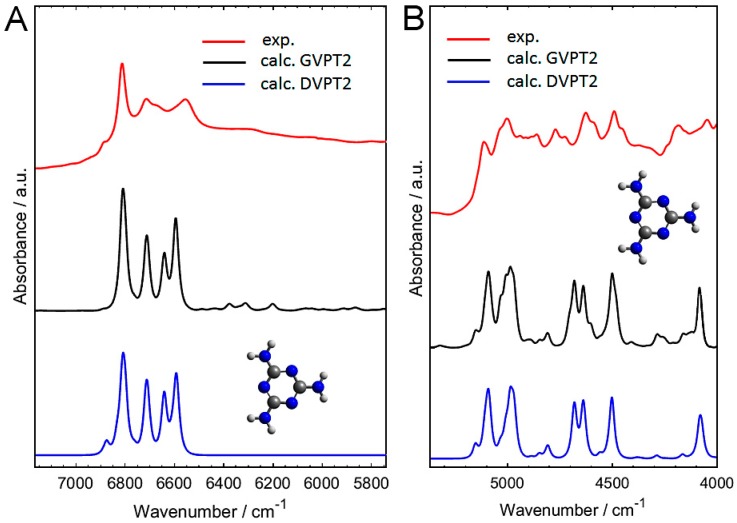
Melamine (IUPAC: 1,3,5-Triazine-2,4,6-triamine) attracts high attention in analytical vibrational spectroscopy due to its misuse as a food adulterant. Vibrational spectroscopy [infrared (IR) and Raman and near-infrared (NIR) spectroscopy] is a major quality control tool in the detection and quantification of melamine content. The physical background for the measured spectra is not interpreted in analytical spectroscopy using chemometrics. In contrast, quantum mechanical calculations are capable of providing deep and independent insights therein. So far, the NIR region of crystalline melamine has not been studied by quantum mechanical calculations, while the investigations of its IR spectra have remained limited. In the present work, we employed fully anharmonic calculation of the NIR spectrum of melamine based on finite models, and also performed IR spectral simulation by using an infinite crystal model-periodic in three dimensions. This yielded detailed and unambiguous NIR band assignments and revised the previously known IR band assignments. We found that the out-of-plane fundamental transitions, which are essential in the IR region, are markedly more sensitive to out-of-plane inter-molecular interactions of melamine than NIR transitions. Proper description of the chemical surrounding of the molecule of melamine is more important than the anharmonicity of its vibrations. In contrast, the NIR bands mostly arise from in-plane vibrations, and remain surprisingly insensitive to the chemical environment. These findings explain previous observations that were reported in IR and NIR analytical studies of melamine.
三聚氰胺(IUPAC:1,3,5-三嗪-2,4,6-三胺)由于被滥用作食品掺杂物而在分析振动光谱学中引起了高度关注。振动光谱学(红外(IR)和拉曼和近红外(NIR)光谱学)是检测和定量三聚氰胺含量的主要质量控制工具。在使用化学计量学的分析光谱学中,未对测量的光谱的物理背景进行解释。相比之下,量子力学计算能够提供深入且独立的见解。到目前为止,尚未通过量子力学计算研究结晶三聚氰胺的近红外区域,而对其红外光谱的研究仍然有限。在本工作中,我们基于有限模型进行了三聚氰胺近红外光谱的全非谐计算,并使用无限晶体模型(在三维上周期性)进行了红外光谱模拟。这得到了详细且明确的近红外带分配,并修正了先前已知的红外带分配。我们发现,在红外区域中必不可少的面外基频跃迁比近红外跃迁对三聚氰胺的面外分子间相互作用更为敏感。三聚氰胺分子的化学环境的适当描述比其振动的非谐性更为重要。相比之下,近红外带主要来自面内振动,并且对化学环境仍然保持惊人的不敏感。这些发现解释了以前在对三聚氰胺进行的红外和近红外分析研究中报告的观察结果。