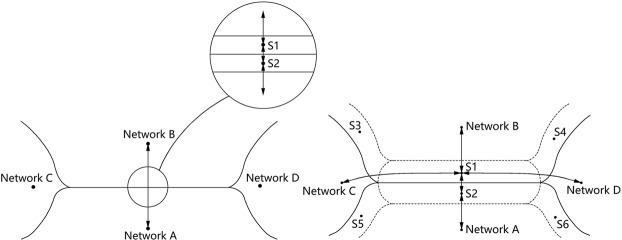
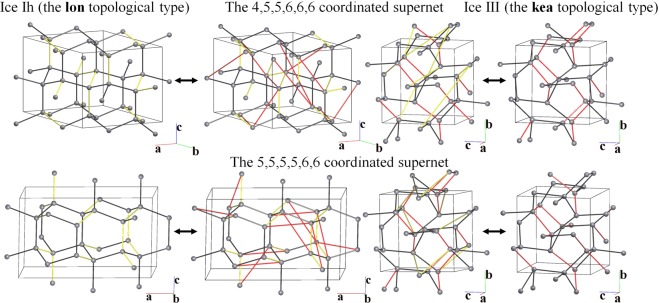
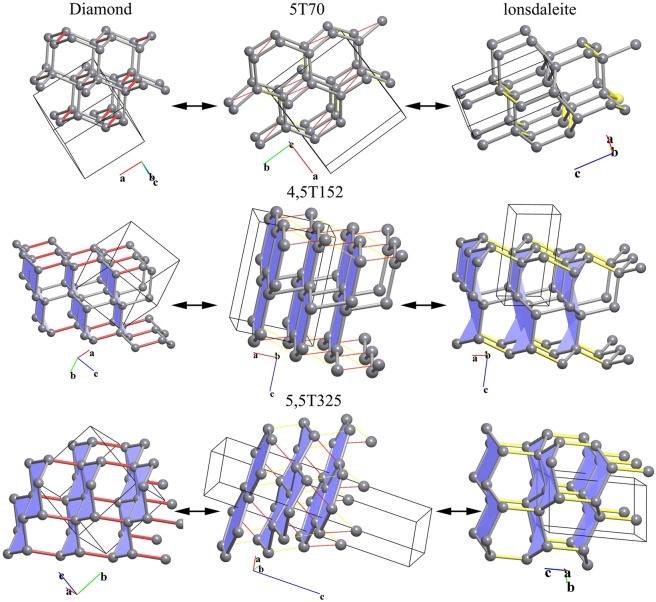
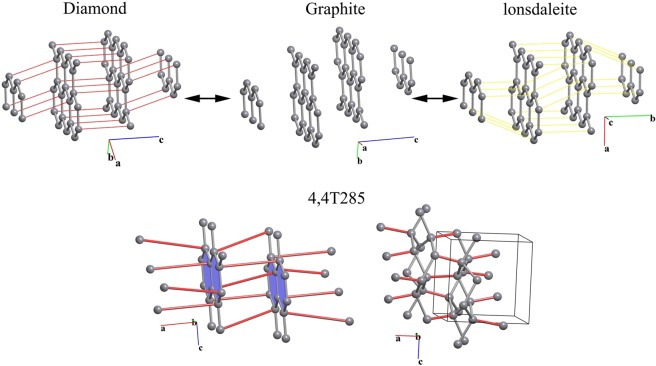
Blatov Vladislav A, Golov Andrey A, Yang Changhao, Zeng Qingfeng, Kabanov Artem A
School of Materials Science and Engineering, Northwestern Polytechnical University, Youyi West Rd. 127, Xi'an, 710072, PR China.
Samara Center for Theoretical Materials Science (SCTMS), Samara State Technical University, Molodogvardeyskaya St. 244, Samara, 443100, Russia.
Sci Rep. 2019 Apr 12;9(1):6007. doi: 10.1038/s41598-019-42483-5.
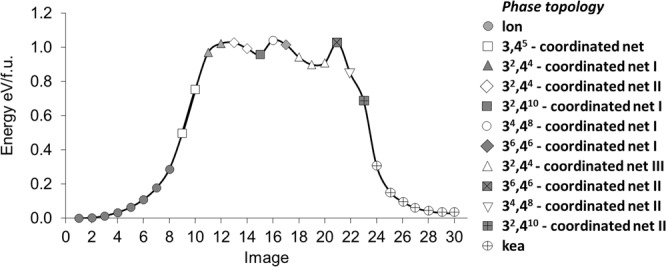
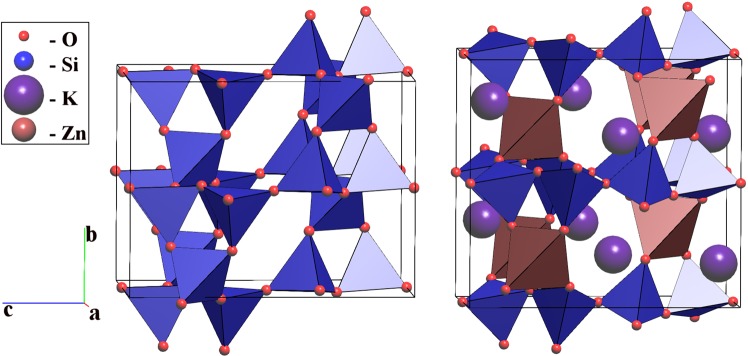
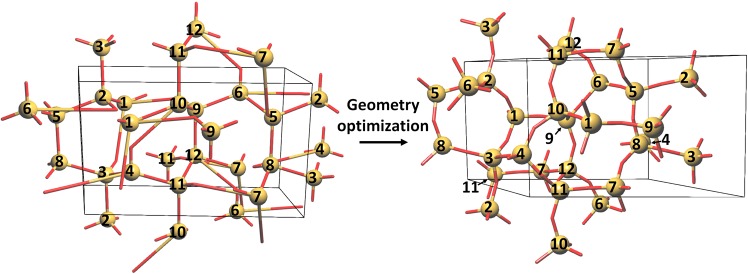
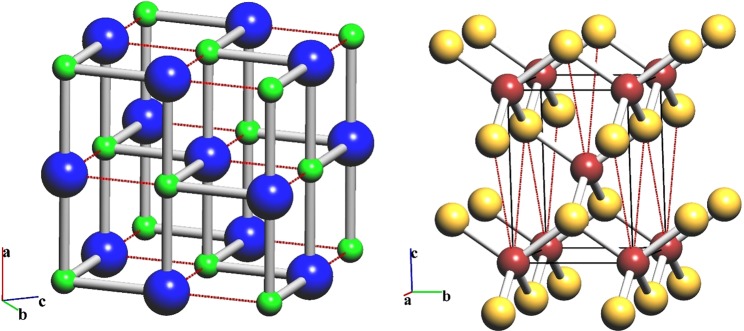
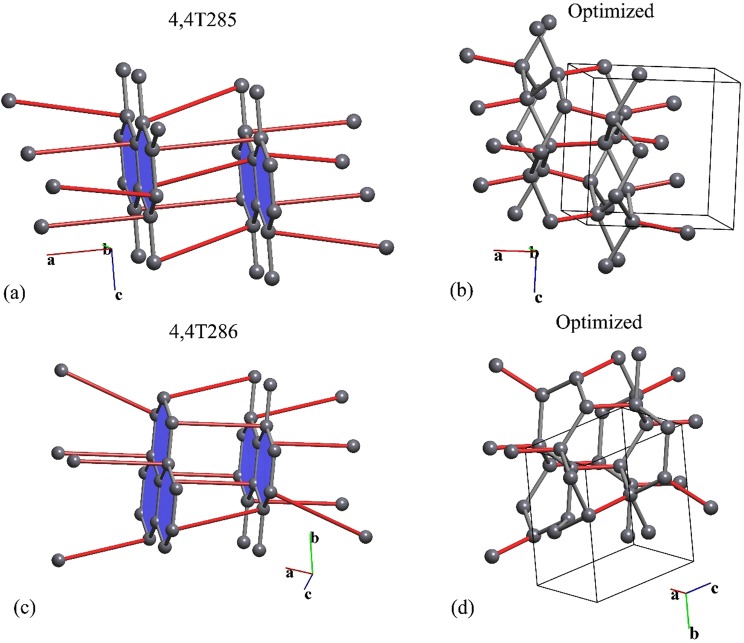
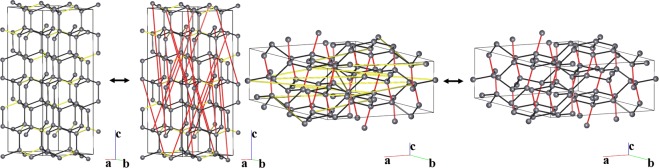
Reconstructive solid-state transformations are followed by significant changes in the system of chemical bonds, i.e. in the topology of the substance. Understanding these mechanisms at the atomic level is crucial for proper explanation and prediction of chemical reactions and phase transitions in solids and, ultimately, for the design of new materials. Modeling of solid-state transitions by geometrical, molecular dynamics or quantum-mechanical methods does not account for topological transformations. As a result, the chemical nature of the transformation processes are overlooked, which limits the predictive power of the models. We propose a universal model based on network representation of extended structures, which treats any reorganization in the solid state as a network transformation. We demonstrate this approach rationalizes the configuration space of the solid system and enables prediction of new phases that are closely related to already known phases. Some new phases and unclear transition pathways are discovered in example systems including elementary substances, ionic compounds and molecular crystals.
重构固态转变之后,化学键系统会发生显著变化,即物质的拓扑结构会发生变化。在原子层面理解这些机制对于正确解释和预测固体中的化学反应及相变至关重要,最终对于新材料的设计也至关重要。用几何方法、分子动力学方法或量子力学方法对固态转变进行建模并未考虑拓扑转变。因此,转变过程的化学本质被忽视了,这限制了模型的预测能力。我们提出了一个基于扩展结构网络表示的通用模型,该模型将固态中的任何重组都视为网络转变。我们证明这种方法使固体系统的构型空间合理化,并能够预测与已知相密切相关的新相。在包括单质、离子化合物和分子晶体的示例系统中发现了一些新相和不明确的转变途径。