Zhang Mingzhen, Jang Hyunbum, Nussinov Ruth
Computational Structural Biology Section , Basic Science Program , Frederick National Laboratory for Cancer Research , Frederick , MD 21702 , USA . Email:
Department of Human Molecular Genetics and Biochemistry , Sackler School of Medicine , Tel Aviv University , Tel Aviv 69978 , Israel.
Chem Sci. 2019 Feb 20;10(12):3671-3680. doi: 10.1039/c8sc04498h. eCollection 2019 Mar 28.
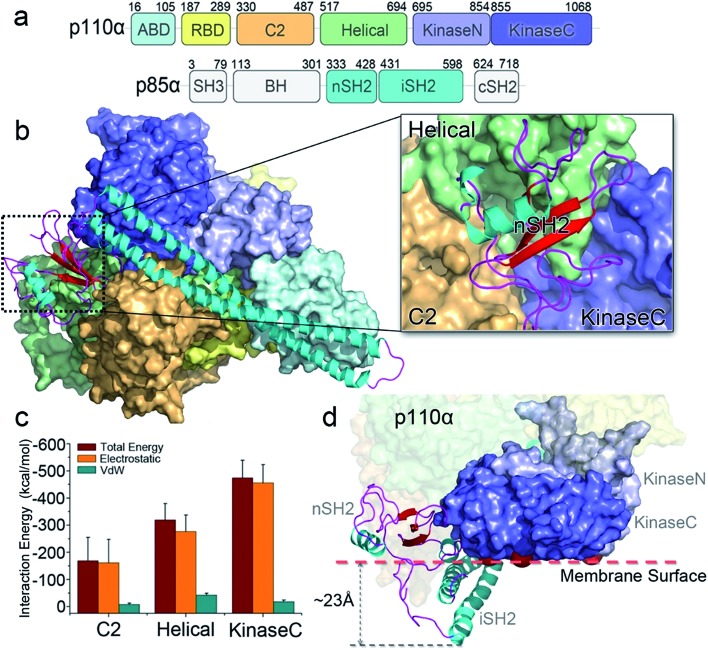
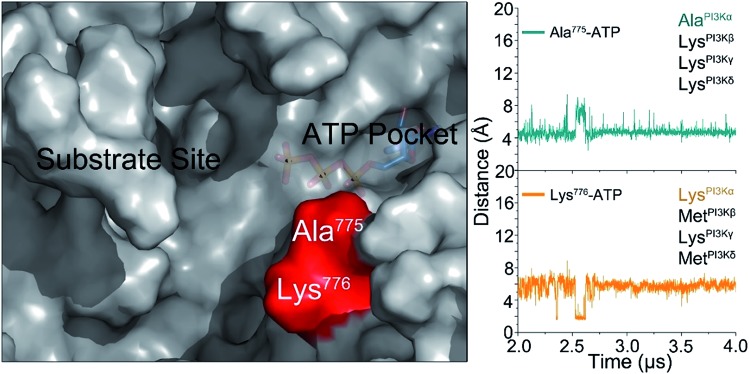
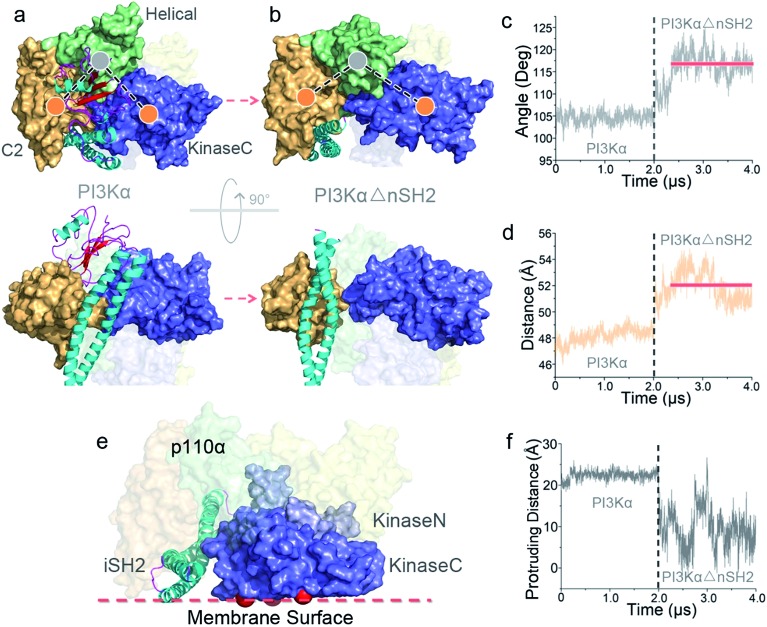
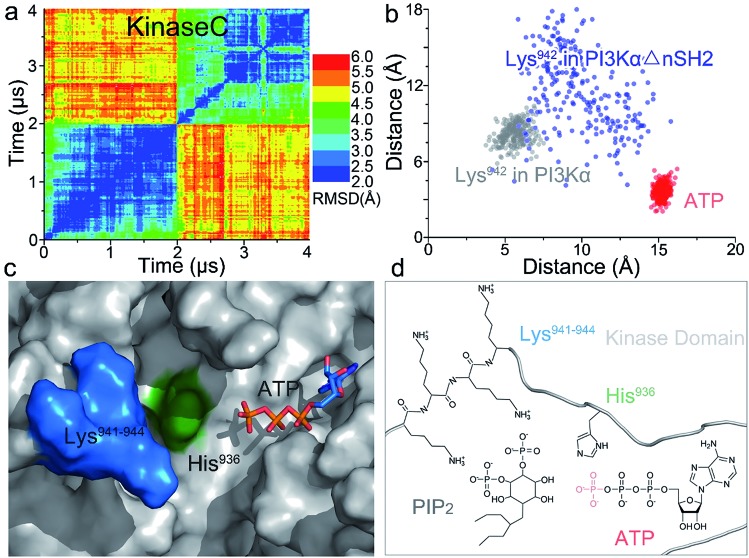
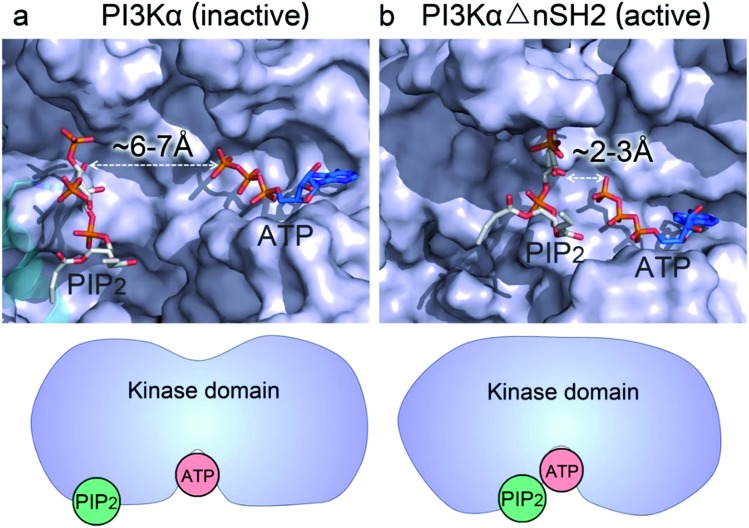
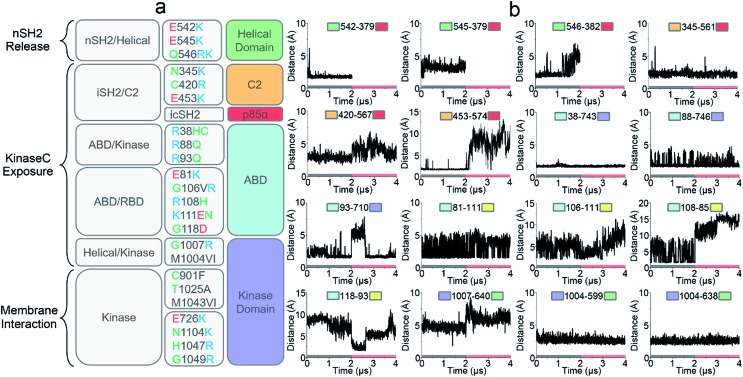
PI3K lipid kinases phosphorylate PIP to PIP in the PI3K/Akt/mTOR pathway to regulate cellular processes. They are frequently mutated in cancer. Here we determine the PI3Kα activation mechanism at the atomic level. Unlike protein kinases where the substrate abuts the ATP, crystal structures indicate that in PI3Kα, the distance between the γ phosphate of the ATP and the PIP lipid substrate is over 6 Å, much too far for the phosphoryl transfer, raising the question of how catalysis is executed. PI3Kα has two subunits, the catalytic p110α and the regulatory p85α. Our simulations show that release of the autoinhibition exerted by the nSH2 domain of the p85α triggers significant conformational change in p110α, leading to the exposure of the kinase domain for membrane interaction. Structural rearrangement in the C-lobe of the kinase domain reduces the distance between the ATP γ-phosphate and the substrate, offering an explanation as to how phosphoryl transfer is executed. An alternative mechanism may involve ATP relocation. This mechanism not only explains how oncogenic mutations promote PI3Kα activation by facilitating nSH2 release, or nSH2-release-induced, allosteric motions; it also offers an innovative, PI3K isoform-specific drug discovery principle. Rather than competing with nanomolar range ATP in the ATP-binding pocket and contending with ATP pocket conservation and massive binding targets, this mechanism suggests blocking the PI3Kα sequence-specific cavity between the ATP-binding pocket and the substrate binding site. Targeting isoform-specific residues in the cavity may prevent PIP phosphorylation.
PI3K脂质激酶在PI3K/Akt/mTOR信号通路中将PIP磷酸化为PIP,以调节细胞过程。它们在癌症中经常发生突变。在此,我们在原子水平上确定了PI3Kα的激活机制。与底物紧邻ATP的蛋白激酶不同,晶体结构表明,在PI3Kα中,ATP的γ磷酸基团与PIP脂质底物之间的距离超过6 Å,对于磷酸转移来说太远了,这就提出了催化是如何进行的问题。PI3Kα有两个亚基,催化性的p110α和调节性的p85α。我们的模拟表明,p85α的nSH2结构域施加的自抑制作用的释放会触发p110α的显著构象变化,导致激酶结构域暴露以进行膜相互作用。激酶结构域C叶中的结构重排减少了ATPγ磷酸基团与底物之间的距离,解释了磷酸转移是如何进行的。另一种机制可能涉及ATP的重新定位。这种机制不仅解释了致癌突变如何通过促进nSH2释放或nSH2释放诱导的变构运动来促进PI3Kα的激活;它还提供了一种创新的、PI3K亚型特异性的药物发现原则。该机制不是在ATP结合口袋中与纳摩尔范围的ATP竞争,也不是与ATP口袋的保守性和大量结合靶点竞争,而是建议阻断ATP结合口袋和底物结合位点之间的PI3Kα序列特异性腔。靶向该腔中的亚型特异性残基可能会阻止PIP磷酸化。