BioForA, INRA, ONF, 45075, Orléans, France, 2163 Avenue de la Pomme de Pin CS 40001 ARDON, Orléans Cedex 2, 45075, France.
Etude du Polymorphisme des Génomes Végétaux (EPGV), INRA, Université Paris-Saclay, 91000, 2 rue Gaston Crémieux, Evry, 9100, France.
BMC Genomics. 2019 Apr 18;20(1):302. doi: 10.1186/s12864-019-5660-y.
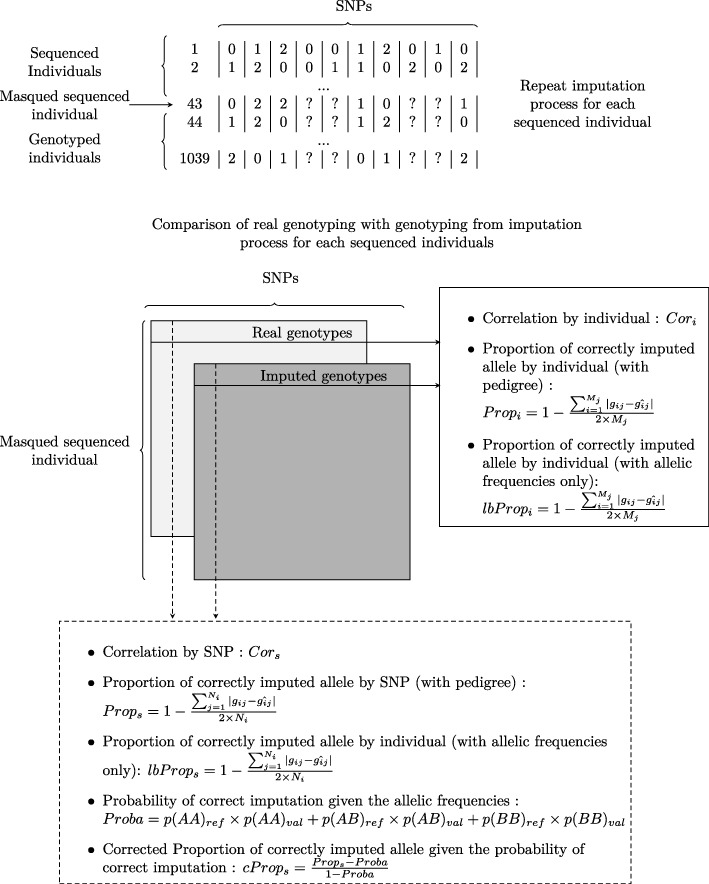
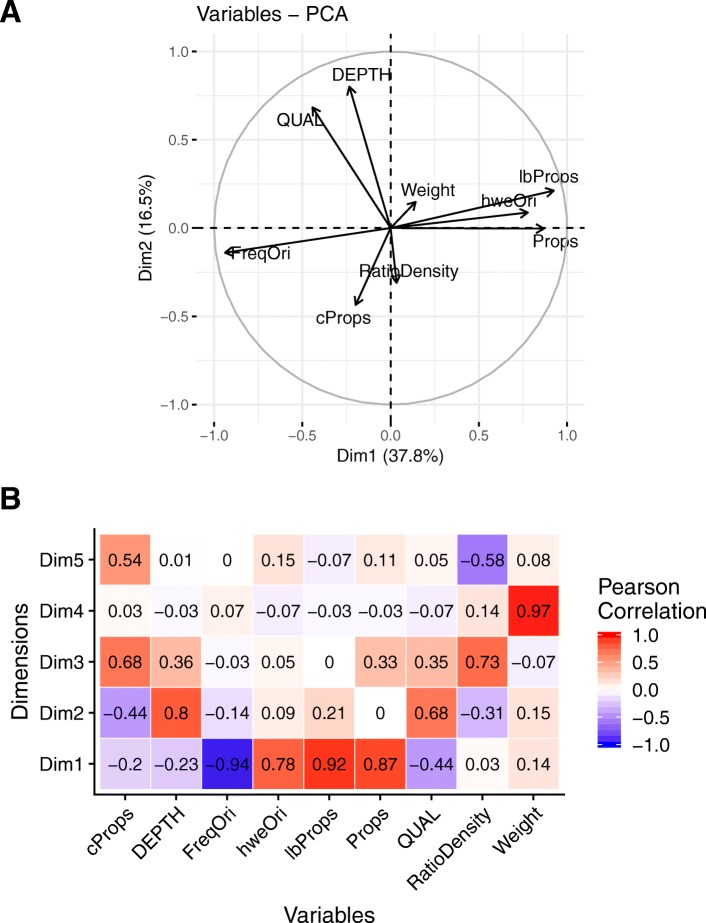
Genomic selection accuracy increases with the use of high SNP (single nucleotide polymorphism) coverage. However, such gains in coverage come at high costs, preventing their prompt operational implementation by breeders. Low density panels imputed to higher densities offer a cheaper alternative during the first stages of genomic resources development. Our study is the first to explore the imputation in a tree species: black poplar. About 1000 pure-breed Populus nigra trees from a breeding population were selected and genotyped with a 12K custom Infinium Bead-Chip. Forty-three of those individuals corresponding to nodal trees in the pedigree were fully sequenced (reference), while the remaining majority (target) was imputed from 8K to 1.4 million SNPs using FImpute. Each SNP and individual was evaluated for imputation errors by leave-one-out cross validation in the training sample of 43 sequenced trees. Some summary statistics such as Hardy-Weinberg Equilibrium exact test p-value, quality of sequencing, depth of sequencing per site and per individual, minor allele frequency, marker density ratio or SNP information redundancy were calculated. Principal component and Boruta analyses were used on all these parameters to rank the factors affecting the quality of imputation. Additionally, we characterize the impact of the relatedness between reference population and target population.
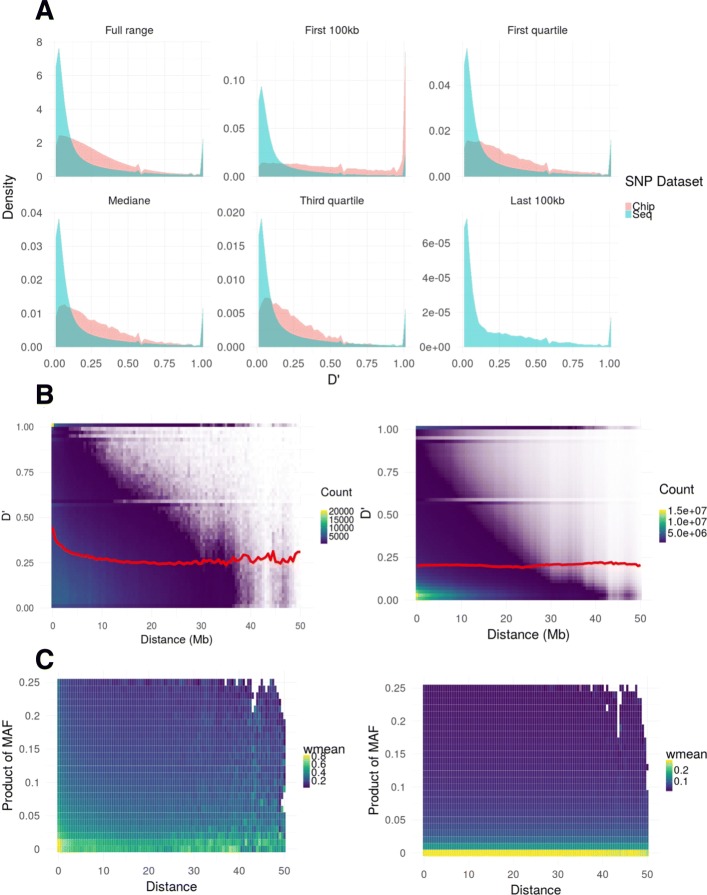
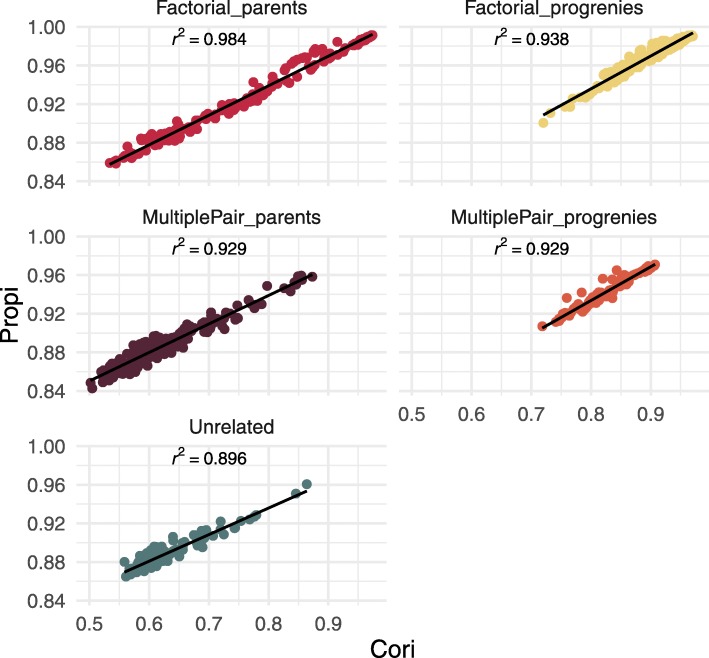
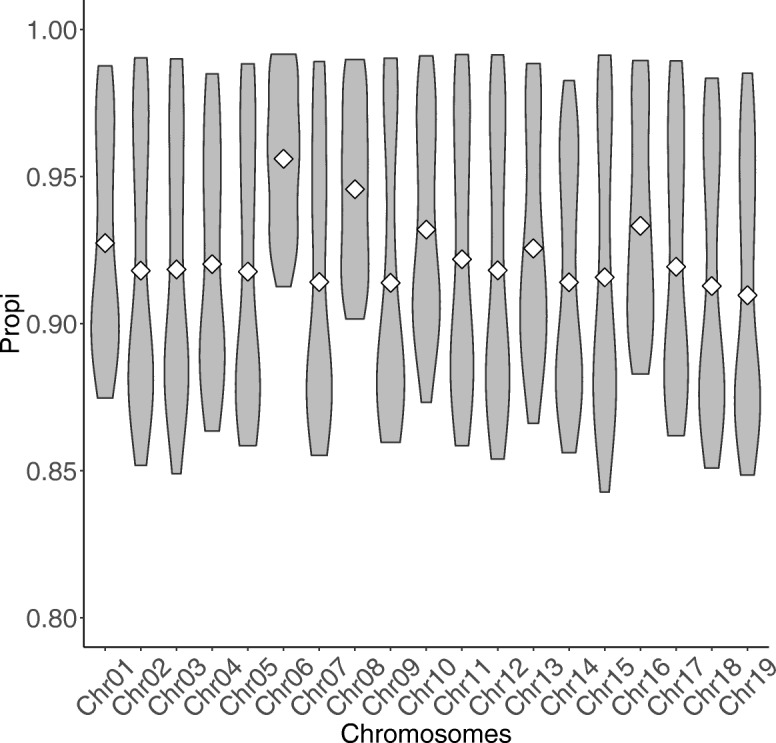
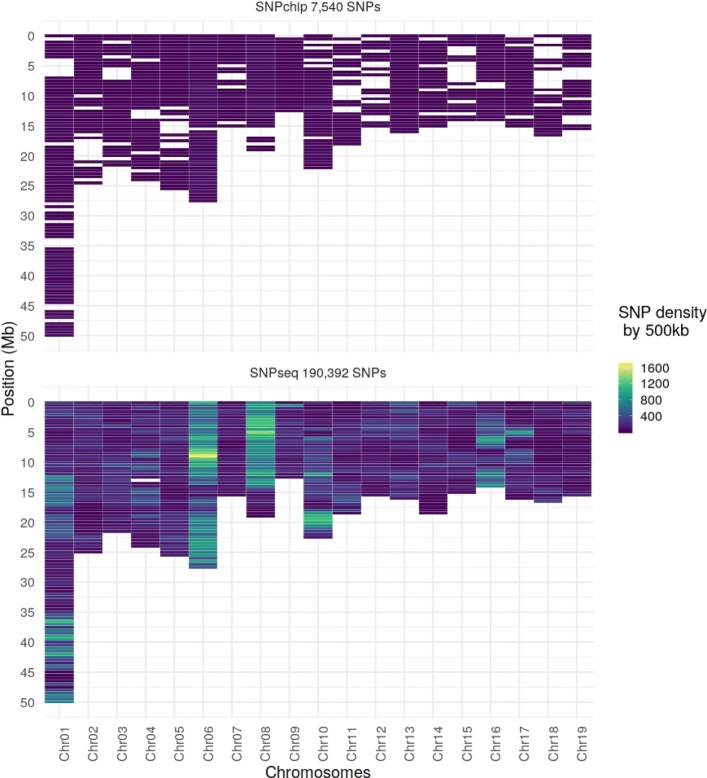
During the imputation process, we used 7540 SNPs from the chip to impute 1,438,827 SNPs from sequences. At the individual level, imputation accuracy was high with a proportion of SNPs correctly imputed between 0.84 and 0.99. The variation in accuracies was mostly due to differences in relatedness between individuals. At a SNP level, the imputation quality depended on genotyped SNP density and on the original minor allele frequency. The imputation did not appear to result in an increase of linkage disequilibrium. The genotype densification not only brought a better distribution of markers all along the genome, but also we did not detect any substantial bias in annotation categories.
This study shows that it is possible to impute low-density marker panels to whole genome sequence with good accuracy under certain conditions that could be common to many breeding populations.
基因组选择的准确性随着 SNP(单核苷酸多态性)覆盖率的提高而提高。然而,这种覆盖范围的增加需要付出高昂的成本,这使得育种者无法迅速实施。在基因组资源开发的早期阶段,低密度面板被推断为更高密度的面板提供了一种更便宜的选择。我们的研究首次探索了树木物种中的推断:黑杨。从一个育种群体中选择了大约 1000 棵纯黑杨,并使用 12K 定制的 Infinium Bead-Chip 进行了基因型分析。其中 43 棵对应于系谱中的节点树的个体被完全测序(参考),而其余的大多数(目标)则通过 FImpute 从 8K 推断到 140 万 SNPs。每个 SNP 和个体都在 43 棵测序树的训练样本中通过留一法交叉验证来评估推断错误。通过 Hardy-Weinberg 平衡精确检验 p 值、测序质量、每个位点和个体的测序深度、次要等位基因频率、标记密度比或 SNP 信息冗余等一些汇总统计数据来计算。对所有这些参数进行主成分和 Boruta 分析,以对影响推断质量的因素进行排名。此外,我们还描述了参考群体和目标群体之间的亲缘关系对推断的影响。
在推断过程中,我们使用芯片上的 7540 个 SNP 来推断来自序列的 1438827 个 SNP。在个体水平上,SNP 的正确推断比例在 0.84 到 0.99 之间,推断精度较高。准确性的变化主要是由于个体之间的亲缘关系不同。在 SNP 水平上,推断质量取决于基因分型 SNP 的密度和原始的次要等位基因频率。推断似乎没有导致连锁不平衡的增加。基因型密集化不仅使标记在整个基因组上的分布更好,而且我们没有检测到注释类别有任何实质性的偏差。
本研究表明,在某些条件下,有可能以较高的准确性将低密度标记面板推断为全基因组序列,这些条件可能在许多育种群体中都很常见。